分子动力学
分子动力学是模拟分子的逐个原子行为并从这些原子运动中推导出宏观属性的技术。它适用于生物分子,如蛋白质和核酸,以及材料科学和纳米技术的天然和合成分子。分子动力学属于粒子方法的范畴,其中包括天体力学和天体物理学中的N体问题,这里介绍的许多观点将延续到这些其他领域。此外,还有一些分子动力学的特殊情况,包括从头开始的分子动力学,其中电子被量子力学地处理,因此可以对化学反应进行建模。我们将不讨论这些特殊情况,而是集中讨论经典的分子动力学。
分子动力学的想法非常简单:一组粒子根据牛顿运动定律相互作用,$F = ma$。考虑到初始粒子的位置和速度、粒子的质量和其他参数,以及粒子之间的作用力模型,牛顿运动定律可以通过数值集成来给出每个粒子在未来(和过去)所有时间的运动轨迹。通常情况下,粒子居住在一个具有周期性边界条件的计算箱中。
因此,一个分子动力学的时间步骤由两部分组成。
计算各粒子的力量
更新位置(整合)。
力的计算是开销最大的部分。最先进的分子动力学模拟是在并行计算机上进行的,因为力的计算十分复杂,而且合理的模拟长度需要大量的时间步骤。在许多情况下,分子动力学也被应用于具有非常多原子的分子的模拟,例如,对于生物分子和长时间尺度来说,可以达到一百万,对于其他分子和短时间尺度来说,可以达到数十亿。
数值整合技术在分子动力学中也很有意义。对于需要大量时间步长的模拟来说,能量等量的保存比准确度更重要,必须使用的求解器与第四章中介绍的传统ODE求解器不同。
在下文中,我们将介绍用于生物分子模拟的力场,并讨论计算这些力的快速方法。然后,我们将专门讨论短程力的分子动力学并行化和用于长程力快速计算的三维 FFT的并行化。
最后,我们用一个章节来介绍适合于分子动力学模拟的集成技术类别。我们在本章中对分子动力学主题的处理是为了介绍和实用;如果想了解更多的信息,建议阅读文本[68]。
受力计算
力场分析
在经典的分子动力学中,势能和原子间作用力的模型被称为力场。力场是一个可操作但近似的量子力学效应模型,对于大分子来说,计算成本太高,无法确定。不同的力场用于不同类型的分子,也有不同的研究人员用于同一分子,而且没有一个是理想的。
在生化系统中,常用的力场将势能函数建模为键合能、范德瓦尔斯能和静电(库仑)能之和。
势是模拟中所有原子位置的函数。原子上的力是原子位置上的这个势的负梯度。粘合能是由于分子中的共价键造成的。
其中,这三个项分别是所有共价键的总和、两个键形成的所有角度的总和以及三个键形成的所有二面体角度的总和。固定参数$k_i, r_{i,0}$等取决于所涉及的原子类型,对于不同的力场可能有所不同。额外的项或具有不同函数形式的项也是常用的。
势能的其余两个项$E$统称为非键合项。理论上,它们构成了力计算的主体。静电能是由原子电荷引起的,其模型是我们熟悉的
其中,总和为所有原子对,$q_i$和$q_j$是原子$i$和$j$的电荷,$r_{ij}$是原子$i$和$j$之间的间距。最后,范德瓦尔斯能近似于剩余的吸引和排斥效应,通常用伦纳德-琼斯函数来模拟
其中$\epsilon_{ij}$和$\sigma_{ij}$是力场参数,取决于原子类型。在短距离上,排斥性的($r^{12}$)项起作用,而在长距离上,分散(吸引,-$r^6$)项起作用。
分子动力学力计算的并行化取决于这些不同类型的力计算的并行化。键合力是局部计算,即对于一个给定的原子,只需要附近的原子位置和数据。范德瓦耳斯力也是局部的,被称为短程的,因为它们对于大的原子分离来说是可以忽略的。静电力是长程的,各种技术已经被开发出来以加快这些计算。在接下来的两个小节中,我们分别讨论短程和长程非键合力的计算。
计算短程非结合力
对一个粒子的短程非粘合力的计算可以在超过该粒子的截止半径$r_c$时被截断。对某一粒子$i$进行这种计算的天真方法是检查所有其他粒子并计算它们与粒子$i$的距离。对于$n$粒子来说,这种方法的复杂性是$O(n^2)$,相当于计算所有粒子对之间的力。有两种数据结构,即单元格列表和Verlet邻居列表,可以独立用于加速这种计算,还有一种方法是将两者结合起来。
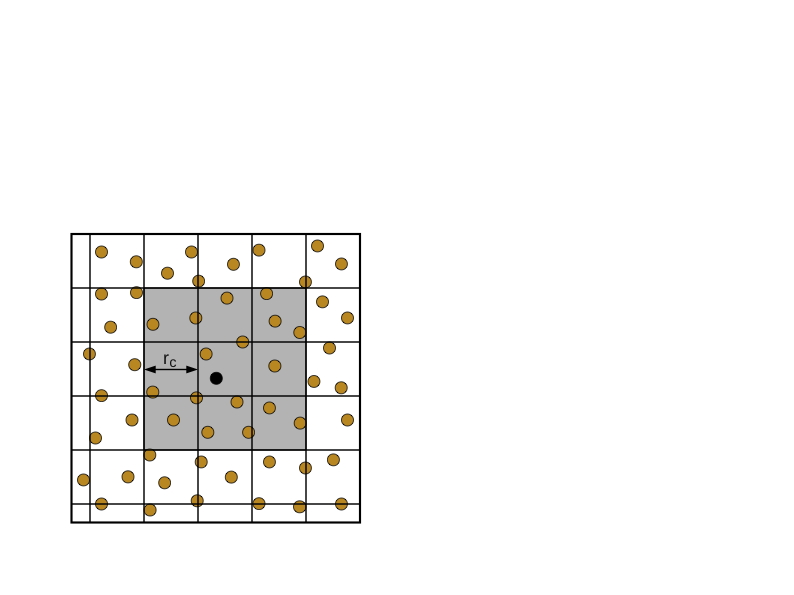
元胞列表
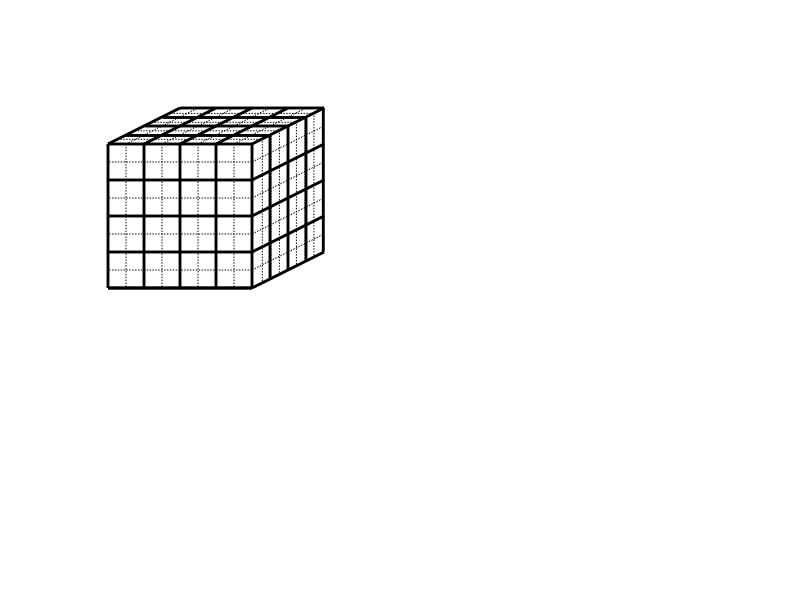
元胞列表的想法经常出现在寻求给定点附近的一组点的问题中。参照图7.1(a),我们用一个二维的例子来说明这个想法,在粒子集合上铺设一个网格。如果网格间距不小于$𝑟_𝑐$,那么为了计算对粒子𝑖的作用力,只需要考虑包含$i$的单元和8个相邻单元的粒子。对所有的粒子进行一次扫描就可以为每个单元构建一个粒子列表。这些元胞列表被用来计算所有粒子的力。在下一个时间步骤中,由于粒子已经移动,必须重新生成或更新元胞列表。这种方法的复杂性是计算数据结构的$O(n)$和计算力的$O(n\times n_c$),其中$n_c$是9个单元的平均粒子数(三维的27个单元)。元胞列表数据结构所需的存储量为$O(n)$。
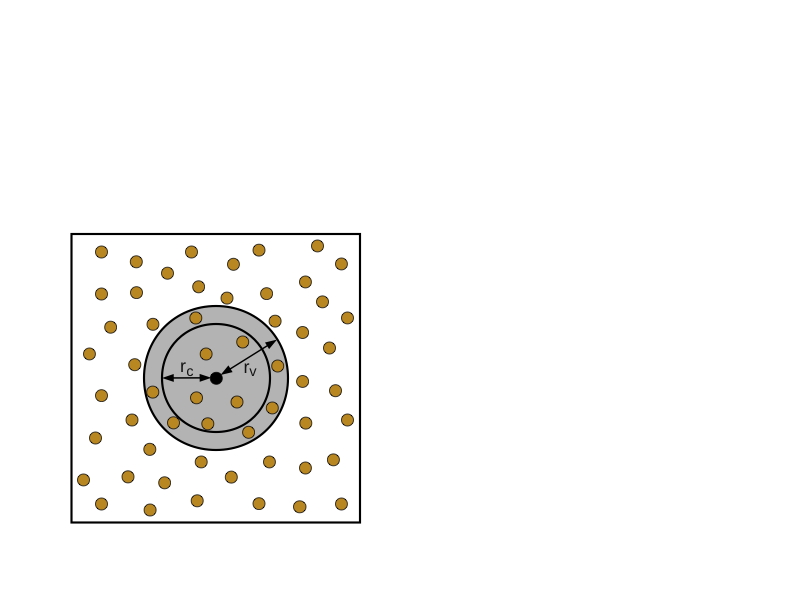
Verlet近邻表
元胞列表结构有些低效,因为对于每个粒子$i$,$n_c$粒子被考虑,但这远远多于截止点$r_c$内的粒子数量。一个Verlet邻居列表是一个粒子𝑖的截止点内的粒子列表。每个粒子都有自己的列表,因此需要的存储量为$O(n\times n_v)$,其中$n_v$是截止点内粒子的平均数量。一旦构建了这些列表,计算力就会非常快,需要最小的复杂度$O(n \times n_v)$。构建列表的成本较高,需要检查每个粒子的所有粒子,即不低于原来的复杂性$O(n^2)$。然而,其优点是,如果使用扩大的截止点$r_v$,近邻表可以在许多时间步骤中重复使用。参考图7.1(b)中的一个二维例子,只要没有来自两个圆圈外的粒子在内圈内移动,近邻表就可以被重复使用。如果粒子的最大速度可以被估计或限定,那么我们可以确定一个时间步数,在这个时间步数内重复使用邻居列表是安全的。(另外,也可以在任何粒子穿越到截止点内的位置时发出信号)。从技术上讲,Verlet近邻表是在扩展的截止点内的粒子列表,$r_v$。
同时使用元胞和近邻表
混合方法是简单地使用Verlet近邻表,但使用元胞列表来构建近邻表。这减少了需要重新生成近邻表时的高成本。这种混合方法非常有效,也是最先进的分子动力学软件中经常使用的方法。
单元列表和Verlet近邻表都可以被修改,以利用以下事实:由于粒子$f_{ij}$的作用力等于$-f_{ji}$(牛顿第三定律),只需要计算一次。例如,对于元胞列表,只需要考虑8个单元格中的4个(在二维)。
计算长程力
静电力的计算具有挑战性,因为它们是长程的:每个粒子都感受到来自模拟中所有其他粒子的不可忽略的静电力。有时使用的一种近似方法是在某个截止半径后截断粒子的力计算(就像对短程范德瓦尔斯力所做的那样)。然而,这通常会在结果中产生不可接受的假象。
有几种更精确的方法可以加快静电力的计算,避免对所有$n$粒子的$O(n^2)$和。我们在此简要介绍其中一些方法。
分层N体方法
分层$N$体方法,包括Barnes-Hut方法和快速多极方法,在天体物理粒子模拟中非常流行,但对于生物分子模拟所要求的精度来说,通常成本太高。在Barnes-Hut方法中,空间被递归地划分为8个相等的单元(三维),直到每个单元包含零或一个粒子。附近的粒子之间的作用力是单独计算的,就像正常情况一样,但是对于远处的粒子,作用力是在一个单元内的一个粒子和一组远处的粒子之间计算的。准确度的测量被用来确定是否可以使用远处的单元来计算力,或者必须通过单独考虑其子单元来计算。Barnes-Hut方法的复杂度为$O(n\log n)$。快速多极法的复杂度为$O(n)$;该方法计算势,不直接计算力。
粒子网格法
在粒子网格方法中,我们利用泊松方程
它将电势$\phi$与电荷密度$\rho$联系起来,其中$1/\epsilon$是一个比例常数。为了利用这个方程,我们用一个网格来离散空间,给网格点分配电荷,解决网格上的泊松方程,得出网格上的势。力是电位的负梯度(对于保守力,如静电力)。许多技术已经被开发出来,用于将空间中的点电荷分布到一组网格点上,也用于数值插值点电荷上由于网格点的电势而产生的力。许多快速方法可用于解决泊松方程,包括多网格方法和快速傅里叶变换。在术语方面,粒子网格方法与原始的粒子-粒子方法相反,后者是在所有粒子对之间计算力。
事实证明,粒子网格法不是很准确,一个更准确的替代方法是将每个力分成短程、快速变化的部分和长程、缓慢变化的部分。
实现这一目标的一个方法是用一个函数$h(r)$来衡量𝑓,它强调短程部分(小$r$用$1-h(r)$来强调长程部分(大$r$)。短程部分是通过计算截止点内所有粒子对的相互作用来计算的(粒子-粒子方法),长程部分是用粒子-网格方法计算的。由此产生的方法,称为粒子-粒子-网格(PPPM,或P3),是由于霍克尼和伊斯特伍德在1973年开始的一系列论文中提出的。
埃瓦尔德法
Ewald方法是迄今为止描述的生物分子模拟中最流行的静电力的方法,是为周期性边界条件的情况而开发的。该方法的结构与PPPM相似,即力被分成短程和长程两部分。同样,短程部分用粒子-粒子方法计算,长程部分用傅里叶变换计算。Ewald方法的变种与PPPM非常相似,因为长程部分使用网格,而快速傅里叶变换被用来解决网格上的泊松方程。更多的细节,请参见,例如[68]。在第7.3节中,我们描述了三维 FFT的并行化以解决三维 Poisson方程。
并行分解
我们现在讨论力的并行计算。Plimpton[169]创建了一个非常有用的分子动力学并行化方法的分类,确定了原子、力和空间分解的方法。在此,我们紧跟他对这些方法的描述。我们还增加了第四类,这类方法已被公认为与前面的类别不同,称为中性领土方法,这个名字是由Shaw[182]创造的。中立领地方法目前被许多最先进的分子动力学代码所使用。空间分解和中性领土方法对基于截止点的计算的并行化特别有利。
原子分解
在原子分解中,每个粒子被分配给一个处理器,该处理器负责计算粒子的力,并在整个模拟中更新其位置。为了使计算大致平衡,每个处理器被分配到大致相同数量的粒子(随机分布效果好)。原子分解的一个重要观点是,每个处理器一般都需要与所有其他处理器进行通信,以共享更新的粒子位置。
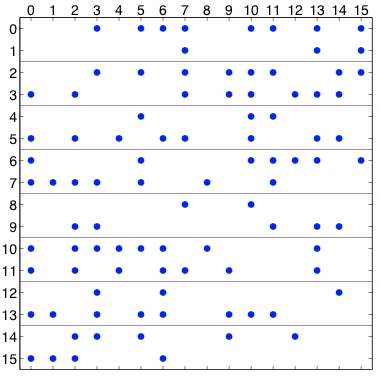
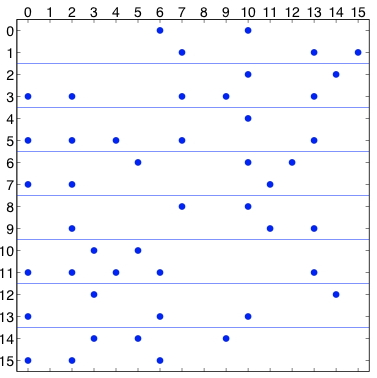
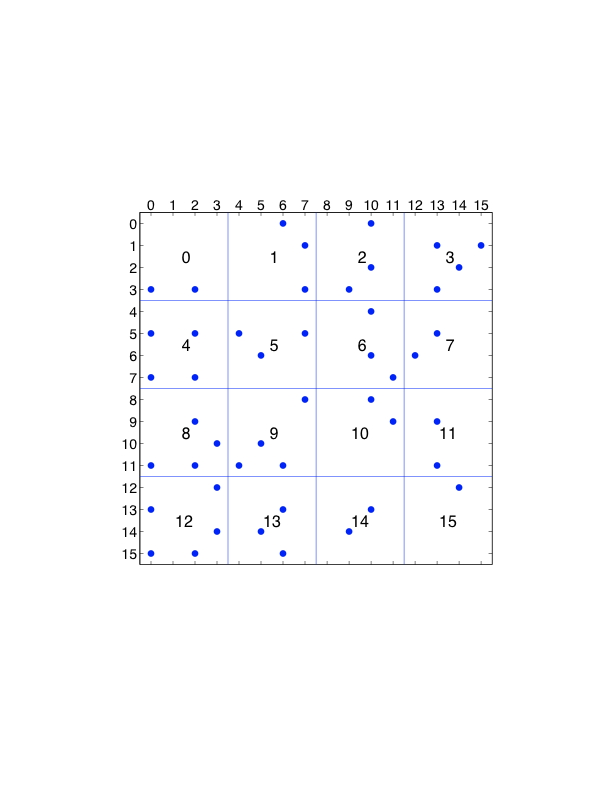
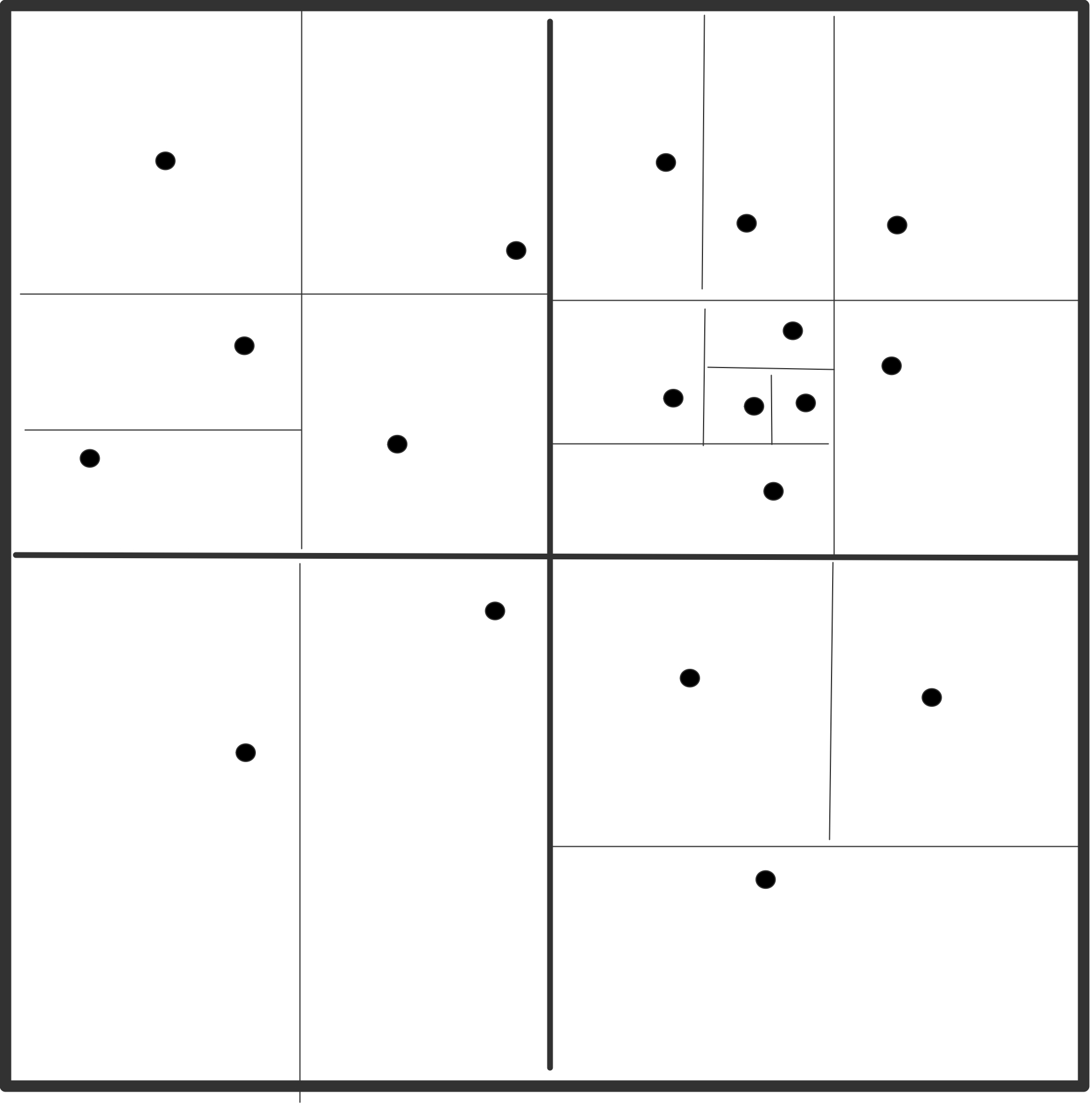
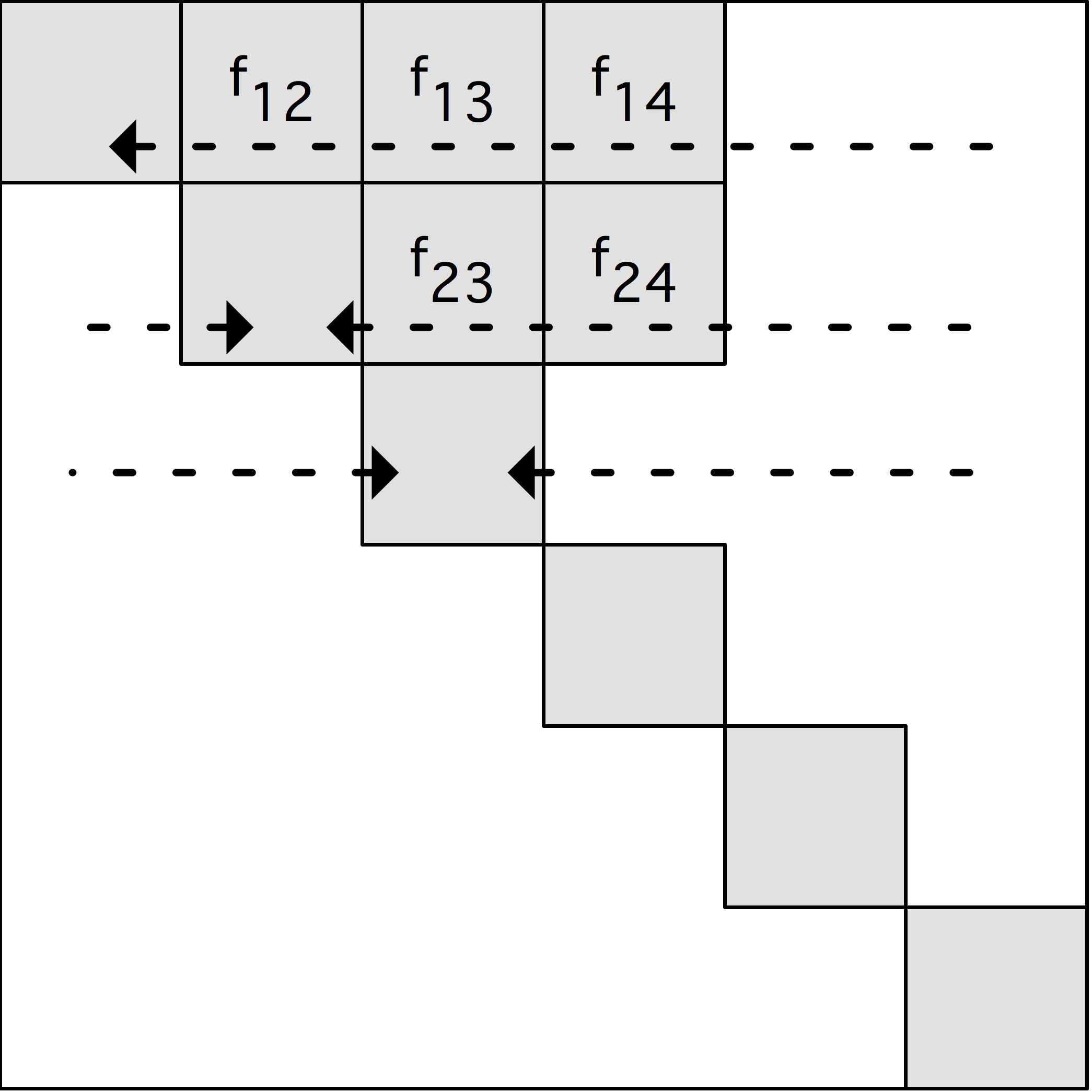
图7.2: 原子分解,显示了分布在8个处理器中的16个粒子的力矩阵。一个点代表力矩阵中的一个非零条目。在左边,矩阵是对称的;在右边,为了利用牛顿第三定律,只计算一对偏斜对称元素中的一个元素。
图7.2(a)中的力矩阵说明了一个原子分解。对于$n$粒子,力矩阵是一个$n\times n$的矩阵;行和列按粒子指数编号。矩阵中的非零条目$f_{ij}$表示由于粒子$j$而对粒子$i$产生的非零力,必须计算出来。这个力可能是一个非粘结力和/或粘结力。当使用截断时,矩阵是稀疏的,如本例中。如果在所有粒子对之间计算力,矩阵是密集的。由于牛顿第三定律$f_{ij}=-f_{ji}$,该矩阵是歪斜对称的。图7.2(a)中的线条显示了粒子是如何被分割的。在图中,16个粒子被划分到8个处理器中。
算法1从一个处理器的角度展示了一个时间步骤。在时间步骤开始时,每个处理器持有分配给它的粒子的位置。
一个优化是将计算量减半,这是有可能的,因为力矩阵是
1 | Algorithm 1 Atom decomposition time step |
歪斜对称的。为了做到这一点,我们为所有偏斜对称对准确选择$f_i$或$f_{ji}$中的一个,这样每个处理器负责计算的力的数量大致相同。选择力矩阵的上三角或下三角部分是一个不好的选择,因为计算负荷是不平衡的。更好的选择是,如果$i + f_{ij}$在上三角中是偶数,或者如果$i + j$在下三角中是奇数,就计算$j$,如图7.2(b)所示。还有许多其他选择。
当利用力矩阵中的歪斜对称性时,一个处理器所拥有的粒子上的所有力不再由该处理器来计算。例如,在图7.2(b)中,粒子1上的力不再只由第一个处理器计算。为了完成力的计算,处理器必须通过通信来发送其他处理器需要的力,并接收其他处理器计算的力。现在必须修改上述算法,增加一个通信步骤(步骤4),如算法2所示。
1 | Algorithm 2 Atom decomposition time step, without redundant calculations 1: send/receiveparticlepositionsto/fromallotherprocessors |
如果额外的通信量被节省的计算量所抵消,这种算法是有利的。请注意,一般情况下,通信量会增加一倍。
力的分解
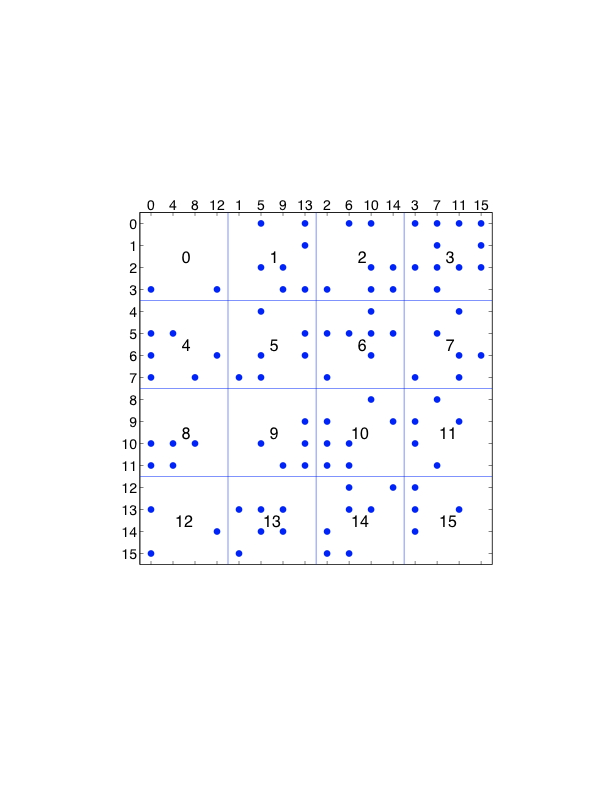
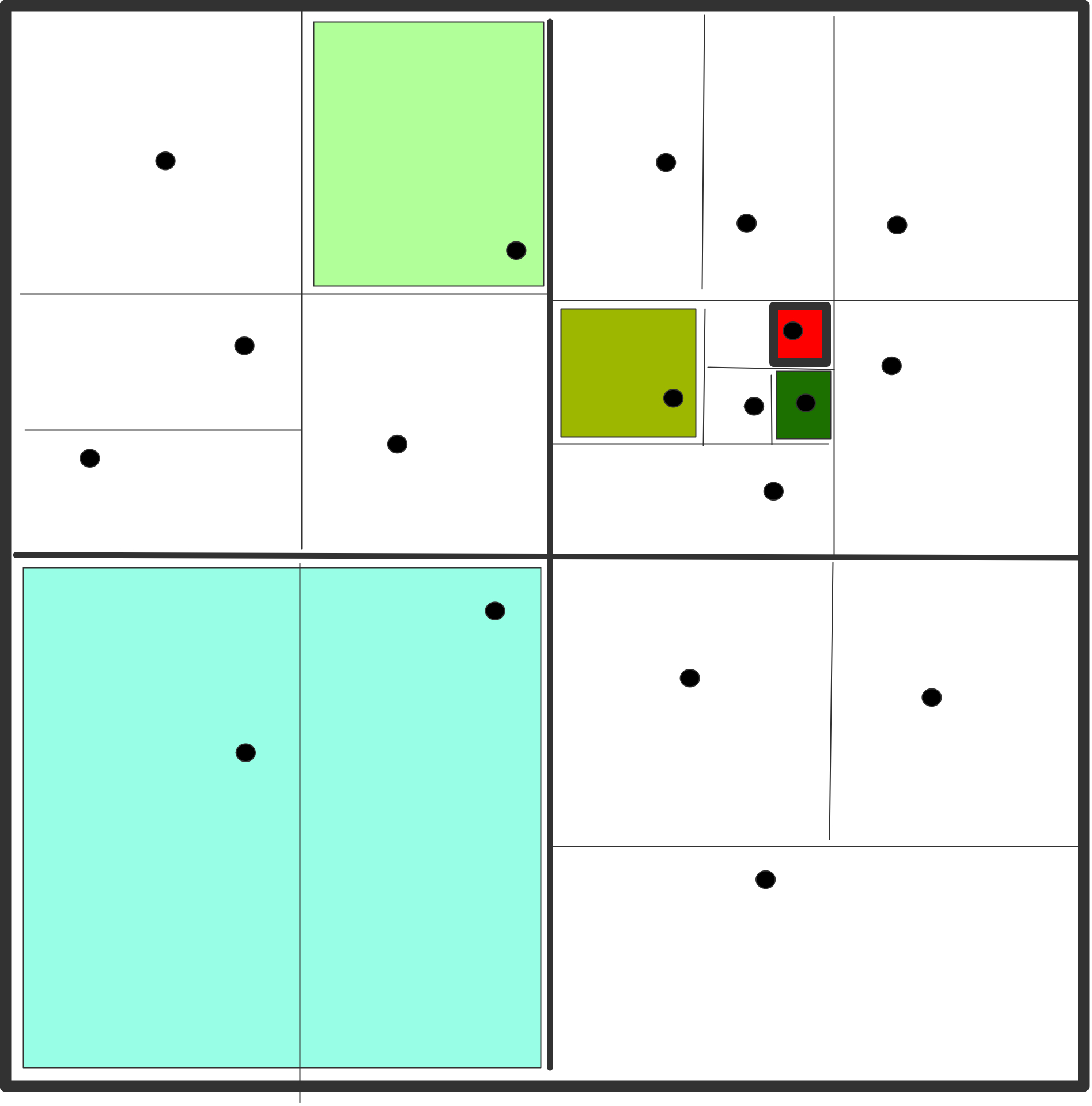
在力的分解中,力被分配到各处理器中进行计算。做到这一点的直接方法是将力矩阵划分为块,并将每个块分配给一个处理器。图7.3(a)说明了16个粒子和16个处理器的情况。粒子也需要被分配给处理器(如在原子分解中),目的是让处理器被分配来更新粒子的位置。在图中的例子中,处理器𝑖被分配来更新粒子𝑖的位置;在实际问题中,一个处理器会被分配来更新许多粒子的位置。请注意,同样,我们首先考虑的是倾斜对称的力矩阵的情况。
现在我们来看看一个时间步骤中力的分解所需的通信。考虑处理器3,它计算粒子0、1、2、3的部分力,并需要粒子0、1、2、3以及12、13、14、15的位置。因此,处理器3需要与处理器0、1、2、3,以及处理器12、13、14、15进行通信。在所有处理器计算完力之后,处理器3需要收集由其他处理器计算的对粒子3的力。因此,处理器2需要再次与处理器0、1、2、3进行通信。
算法3显示了从一个处理器的角度来看,在一个时间步骤中所进行的工作。在时间步骤开始时,每个处理器持有分配给它的所有粒子的位置。
1 | Algorithm 3 Force decomposition time step |
一般来说,如果有$p$个处理器(为简单起见,$p$为正方形),那么力矩阵被$\sqrt{p}$个块分割成$\sqrt{p}$个。刚刚描述的力的分解需要处理器分三步进行通信,每一步都有$\sqrt{p}$个处理器。这比需要在所有$p$处理器之间进行通信的原子分解要有效得多。
我们还可以利用牛顿第三定律进行力的分解。像原子分解一样,我们首先选择一个修改过的力矩阵,其中只有$f_i$和$f_{ji}$中的一个被计算。粒子$i$上的力是由一排处理器计算的,现在也由一列处理器计算。因此,每个处理器需要一个额外的通信步骤,从一列处理器中收集分配给它的粒子的力。以前有三个通信步骤,现在利用牛顿第三定律时有四个通信步骤(在这种情况下,通信不是像原子分解那样加倍的)。
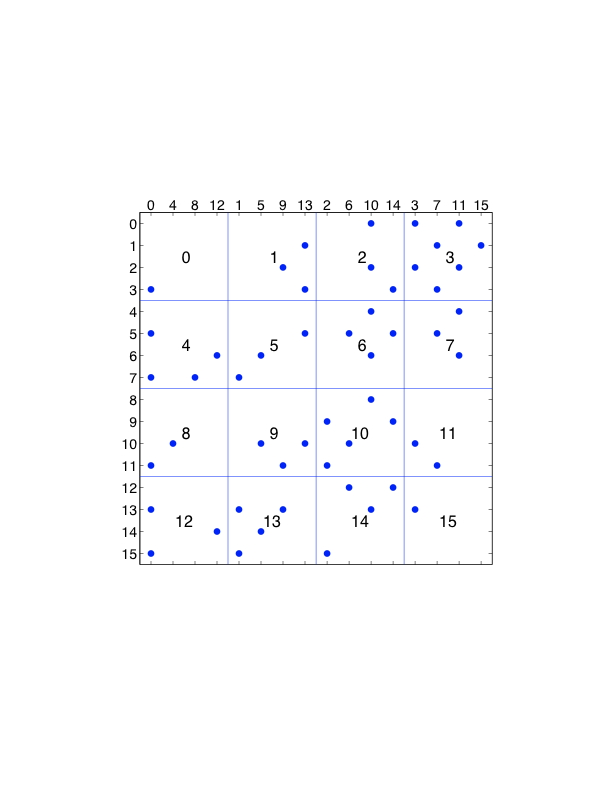
对力的分解的修改可以节省一些通信。在图7.4中,各列被重新排序,使用的是块-循环排序。再考虑一下处理器3,它计算粒子0、1、2、3的部分力。它需要来自粒子0、1、2、3的位置,和以前一样,但现在也需要处理器3、7、11、15。后者是与处理器3同列的处理器。因此,所有通信都在同一处理器行或处理器列内,这在基于网状的网络架构上可能是有利的。修改后的方法显示为算法4。
空间分解
在空间分解中,空间被分解为单元。每个单元被分配给一个处理器,负责计算位于该单元内的粒子上的力。图7.5(a)说明了在二维模拟的情况下,空间被分解为64个单元。(这是空间的分解,不能与力矩阵相混淆)。通常情况下,单元的数量被选择为与处理器的数量相等。由于粒子在模拟过程中移动,粒子在单元中的分配也随之改变。这与原子和力的分解相反。
图7.5(b)显示了一个单元(中间的正方形)和包含可能在截止半径𝑟𝑐内的粒子的空间区域(阴影),这些粒子在给定的单元中。阴影区域通常被称为导入区域,因为给定单元必须导入位于该区域的粒子的位置来进行受力计算。请注意,并不是给定单元中的所有粒子都必须与导入区域中的所有粒子相互作用,特别是如果导入区域与截止半径相比很大的话。
1 | Algorithm 4 Force decomposition time step, with permuted columns of force matrix |
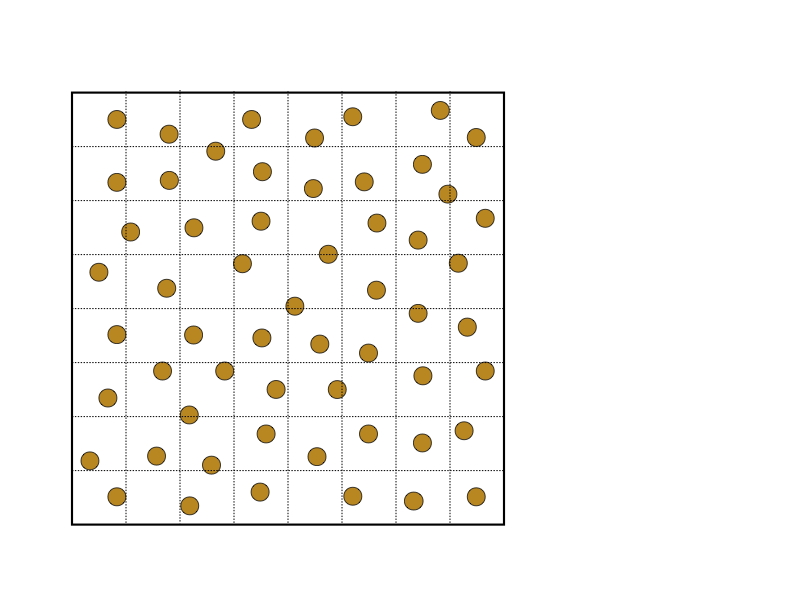
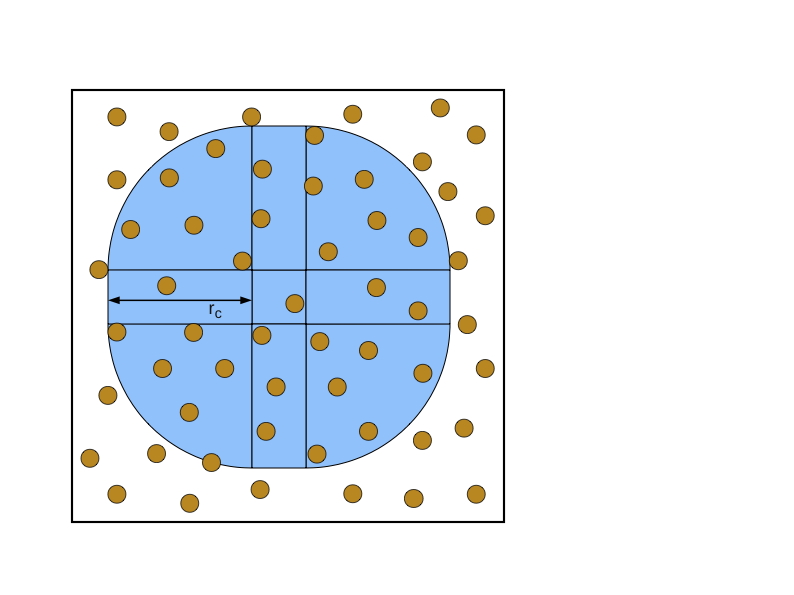
图7.5:空间分解,显示了二维计算盒中的粒子,(a)划分为64个单元,(b)一个单元的导入区域。
算法5显示了每个处理器在一个时间步骤中的执行情况。我们假设在时间步骤开始时,每个处理器持有其单元中的粒子的位置。
为了利用牛顿第三定律,进口区域的形状可以减半。现在,每个处理器只计算其单元中的粒子的部分力,并需要接收来自其他处理器的力来计算这些粒子的总力。因此涉及到一个额外的通信步骤。我们把这个问题留给读者去解决修改后的导入区域和这种情况下的伪代码的细节。
在空间分解方法的实施中,每个单元与它的导入区域的粒子列表相关联,类似于Verlet邻居列表。与Verlet邻居列表一样,如果进口区域略有扩大,则不必在每个时间步骤中更新该列表。这使得进口区域列表可以在几个时间步中重复使用,这与粒子穿越扩展区域的宽度所需的时间是一致的。这与Verlet邻居列表完全类似。
总之,空间分解方法的主要优点是它们只需要在对应于附近粒子的处理器之间进行通信。空间分解方法的一个缺点是,对于非常多的处理器来说,与每个单元内包含的粒子数量相比,导入区域很大。
1 | Algorithm 5 Spatial decomposition time step |
Neutral Territory Methods
我们对Neutral Territory Methods方法的描述与Shaw[182]的描述非常接近。Neutral Territory Methods可以被视为结合了空间分解和力分解的各个方面。为了并行化集成步骤,粒子根据空间的划分被分配到处理器。为了使力的计算并行化,每个处理器计算两组粒子之间的力,但这些粒子可能与被分配给处理器进行整合的粒子没有关系。由于这种额外的灵活性,Neutral Territory Methods需要的通信量可能比空间分解方法少得多。
图7.6是一个二维模拟的Neutral Territory Methods的例子。在图中所示的方法中,给定的处理器被指派计算位于横条的粒子与位于竖条的粒子之间的力。因此,这两个区域构成了这个方法的导入区域。通过与图7.6(b)的比较,这种中性领土方法的导入区域比相应的空间分解方法的导入区域小很多。当每个处理器所对应的单元的大小与截止半径相比很小时,这种优势就更大了。
在计算出力之后,给定的处理器将其计算出的力发送给需要这些力进行整合的处理器。因此,我们有了算法6。
1 | Algorithm 6 Neutral territory method time step |
像其他方法一样,Neutral Territory Methods的进口区域可以被修改以利用牛顿第三定律。我们参考Shaw [182],以了解更多的细节和中立领土方法在三维模拟中的图示。
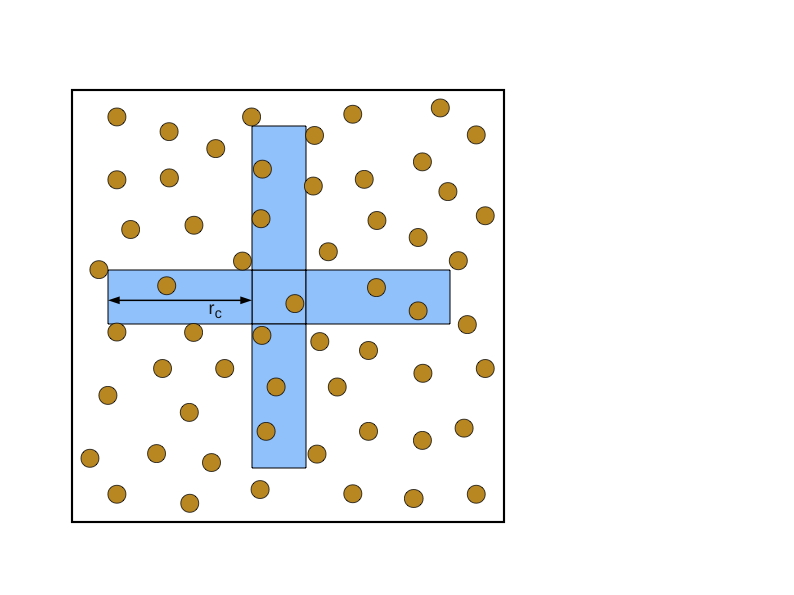
并行快速傅立叶变换
许多计算长程力的方法的一个共同组成部分是用于解决三维网格上泊松方程的三维FFT。傅里叶变换将泊松算子(称为拉普拉斯)对角化,在求解中需要一个正向和一个反向的FFT变换。考虑离散拉普拉斯算子$L$(具有周期性边界条件)和$\phi$在$-L\phi-\rho$的解。让$F$表示傅里叶变换。原问题相当于
矩阵$FLF^{-1}$是对角线。对$\rho$进行正向傅里叶变换$F$,然后用对角线矩阵的逆值对傅里叶空间分量进行缩放,最后,应用反傅里叶变换$F^{-1}$,得到解$\phi$。
对于现实中的蛋白质大小,通常使用大约1埃斯特朗的网格间距,从而导致一个按许多标准来说相当小的三维网格。 64×64×64,或128×128×128。 并行计算通常不会应用于这种大小的问题,但必须使用并行计算,因为数据𝜌已经分布在并行处理器之间(假设使用了空间分解)。 三维FFT是通过沿三个维度依次计算一维FFT来计算的。 对于64×64×64的网格大小,这就是4096个64维的一维FFT。 并行FFT计算通常受到通信的约束。 FFT的最佳并行化取决于变换的大小和计算机网络的结构。 下面,我们首先描述平行一维FFT的一些概念,然后描述平行三维FFT的一些概念。 对于目前致力于大型一维变换的并行化和高效计算(使用SIMD操作)的软件和研究,我们参考了SPIRAL和FFTW软件包。 这些软件包使用自动调谐来生成对用户的计算机结构来说是有效的FFT代码。
并行一维FFT
无转置的一维FFT
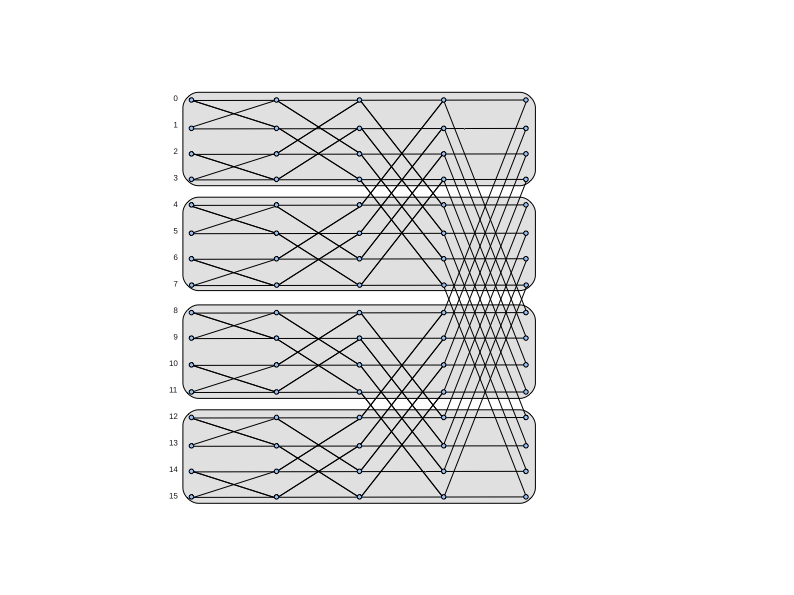
图7.7显示了16点radix-2十进频FFT算法的输入(左边)和输出(右边)之间的数据依赖关系(数据流图)。(图中未显示的是计算中可能需要的位反转变换)。图中还显示了计算在四个处理器之间的划分。在这种并行化中,初始数据不在处理器之间移动,但在计算过程中会发生通信。在图中所示的例子中,通信发生在前两个FFT阶段;最后两个阶段不涉及通信。当通信发生时,每个处理器正好与另外一个处理器进行通信。
转置的一维FFT
使用转置的方式来实现FFT计算的并行化是很常见的。图7.8(a)显示了与图7.7相同的数据流图,但为了清晰起见,删除了水平线并增加了额外的索引标签。和以前一样,前两个FFT阶段是在没有通信的情况下进行的。然后,数据在各处理器之间进行转置。有了这个转置的数据布局,最后两个FFT阶段可以在没有通信的情况下进行。最终的数据不按原来的顺序排列;可能需要额外的转置,或者数据可以按这个转置的顺序使用。图7.8(b)显示了转置前后指数在四个处理器中的分布情况。从这两个图中可以看出,前两个阶段的数据依赖性只涉及同一分区的指数。转置后的后两个阶段的分区情况也是如此。还要注意的是,转置前后的计算结构是相同的。
并行三维FFT
块状分解的三维FFT
图7.9(a)显示了当使用空间分解时,FFT输入数据的块状分解,其大小为8×8×8,分布在以4×4×4拓扑结构排列的64个处理器中。并行一维FFT算法可以在每个维度上应用。对于图中所示的例子,每个一维FFT计算涉及4个处理器。每个处理器同时执行多个一维FFT(本例中为四个)。在每个处理器中,如果穿越其中一个维度,数据是连续排列的,因此在其他两个维度的计算中,数据访问是分层的。分层数据访问可能很慢,因此在计算每个维度的FFT时,可能值得在每个处理器中重新排序数据。
三维FFT与板块分解
图7.9(b)是4个处理器情况下的板块分解图。每个处理器持有输入数据的一个或多个平面。如果输入数据已经分布在板块中,或者可以以这种方式重新分布,就可以使用这种分解。板块平面内的两个一维FFT不需要通信。其余的一维FFT需要通信,可以使用上述两种平行一维FFT的方法之一。板块分解的缺点是,对于大量的处理器来说,处理器的数量可能会超过三维FFT中沿任何一个维度的点的数量。另一个选择是下面的铅笔分解。
三维FFT与铅笔式分解
图7.9(c)显示了16个处理器的情况下的铅笔分解情况。每个处理器持有一个或多个输入数据的铅笔。如果原始输入数据像图7.9(a)那样分布在块中,那么一排处理器之间的通信(在一个三维处理器网中)可以将数据分布到铅笔分解中。然后可以在没有通信的情况下执行一维FFT。为了在另一个维度上执行一维FFT,数据需要被重新分配到另一个维度的铅笔中。在整个三维FFT计算中,总共需要四个通信阶段。
分子动力学的整合
为了对分子动力学中的常微分方程系统进行数值积分,需要采用特殊的方法,与第四章中研究的传统ODE求解器不同。这些特殊的方法被称为对称方法,在产生具有恒定能量的解方面比其他方法更好,例如,对于那些被称为哈密尔顿的系统(包括分子动力学中的系统)。当哈密顿系统在很长的时间间隔内以许多时间步长进行积分时,保留结构(如总能量)往往比方法的精度顺序更重要。在这一节中,我们激励了一些想法,并给出了Störmer-Verlet方法的一些细节,该方法足以用于简单的分子动力学模拟。
哈密顿系统是一类保存能量的动力系统,它可以被写成一种叫做哈密顿方程的形式。为了对称起见,考虑一下简单的谐波振荡器
其中$u$是单个粒子从平衡点的位移。这个方程可以模拟一个具有单位质量的粒子在一个具有单位弹簧常数的弹簧上。处于$u$位置的粒子受到的力是$-u$。这个系统看起来并不像一个分子动力学系统,但对说明几个观点很有用。
上述二阶方程可以写成一阶方程系统
其中$q=u, p=u’$,这是经典力学中常用的符号。一般解决方案是
简谐振荡器的动能是$p^2/2$,势能是$q^2/2$(势能的负梯度是力,$-q$)。因此,总能量与$q^2+p^2$成正比。
现在考虑用三种方法解决一阶方程组,显式欧拉法、隐式欧拉法和一种叫做Störmer-Verlet的方法。 初始条件是$(q, p)$=(1, 0)。 我们使用$h=0.05$的时间步长,走500步。 我们将$q$和$p$分别绘制在横轴和纵轴上(称为相位图)。 如上所述,准确的解决方案是一个以原点为中心的单位圆。 图7.10显示了这些解。 对于显式欧拉,解决方案是向外螺旋式的,这意味着解决方案的位移和动量随时间而增加。 隐式欧拉方法的情况则相反。 总能量的图表将显示这两种情况下的能量分别增加和减少。 当采取较小的时间步长或使用高阶方法时,解决方案会更好,但这些方法完全不适合在长时间内对共轭系统进行积分。 图7.10(c)显示了使用一种叫做Störmer-Verlet方法的对称方法的解。 该解显示,$q^2+p^2$的保存情况比其他两种方法要好得多。
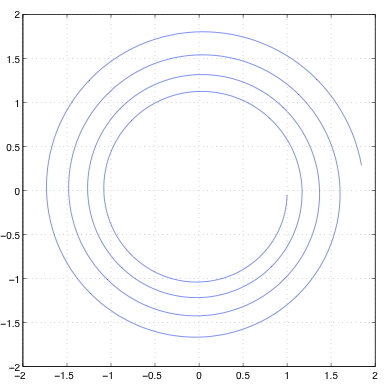
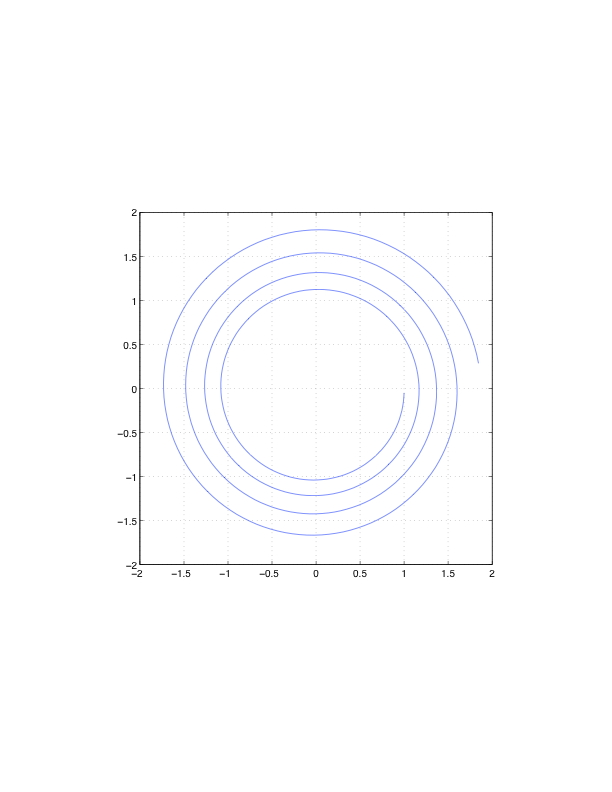
图7.10:初值(1,0)、时间步长0.05、步长500的三种方法的简谐振荡器解的相位图。对于显式Euler,解是向外旋的;对于隐式Euler,解是向内旋的;总能量在Störmer-Verlet方法中保存得最好。 我们推导出二阶方程的Störmer-Verlet方法
通过简单地用有限差分近似值取代左手边
这可以重新排列,得到的方法是
该公式可以等效地从泰勒级数中导出。该方法与线性多步骤方法类似,需要一些其他技术来提供该方法的初始步骤。该方法也是时间可逆的,因为如果$k+1$和$k-1$互换,公式是一样的。遗憾的是,要解释为什么这种方法是对称性的,已经超出了本介绍的范围。
上面写的方法有一些缺点,最严重的是小$h^2$项的添加会受到灾难性的取消。因此,这个公式不应该以这种形式使用,已经开发了一些数学上等效的公式(可以从上面的公式推导出来)。
一个替代公式是跃迁法。
其中𝑣是第一导数(速度),与位移𝑢相差半步。这个公式没有同样的四舍五入问题,而且还提供了速度,尽管它们需要与位移重新对中,以计算给定步骤的总能量。这对方程中的第二个基本上是一个有限差分公式。
Störmer-Verlet方法的第三种形式是速度Verlet变体。
其中,现在速度是在与位移相同的点上计算的。这些算法中的每一种都可以被实现,从而只需要存储两组量(两个先前的位置,或者一个位置和一个速度)。Störmer-Verlet方法的这些变体由于其简单性而受到欢迎,每一步只需要一个昂贵的力评估。 高阶方法通常不实用。 速度Verlet方案也是分子动力学的多时间步长算法的基础。 在这些算法中,慢速变化的(典型的长程)力的评估频率较低,更新位置的频率也比快速变化的(典型的短程)力低。 最后,许多最先进的分子动力学集成了一个经过修改的哈密尔顿系统,以控制模拟温度和压力。 对于这些系统,已经开发出了复杂得多的对称性方法。
组合算法
本部分我们将简要考虑一些组合算法,如:排序算法以及埃拉托色尼筛选法寻找素数法等等。
在科学计算中,排序并不常见,人们通常在数据库(无论是金融数据库还是生物数据库)中考虑排序。当考虑如自适应网格加密(Adaptive Mesh Refinement,AMR)和其他对数据结构的改进中,排序才会凸显其重要之处。
在这一节中,我们将简要介绍一些基本的算法,以及其并行表达。更多的细节,见[131]和其中的参考文献。
排序算法概要
复杂度
众多排序算法以其计算复杂度来区分:即给定$n$元数组,我们需要对它操作多少次能完成排序。
部分排序算法可以达到的最低复杂度为:$O(n\log n)$,例如快速排序(Quicksort)的最佳情况即为$O(n\log n)$。但由于快速排序算法由于需要选择“基准元素”,如果每次选择的基准元素都是最差的,那么其时间复杂度可以达到$O(n^2)$。
1 | while the input array has length > 1 do |
另一方面,非常简单的冒泡排序(bubble sort)算法由于其静态结构,因此时间复杂度恒定。
1 | for pass from 1 to 𝑛 − 1 do |
很容易看出,这个算法的复杂度为$O(n^2)$:内循环做了$t$的比较和最多$t$的交换。从1到$n-1$相加,可以得到大约$n^2/2$的比较和最多相同数量的交换。
排序网络
上面我们看到,部分算法的复杂度不受输入数据的影响,而有些算法则并非如此。前者被称为排序网络(sorting network),可以认为排序网络是实现算法的专用硬件,其基本元件为比较、交换元素。它有两个输入$x$,$y$;两个输出$\max(x,y)$与$\min(x,y)$。
下图展示了由比较和交换元素构成的冒泡排序。
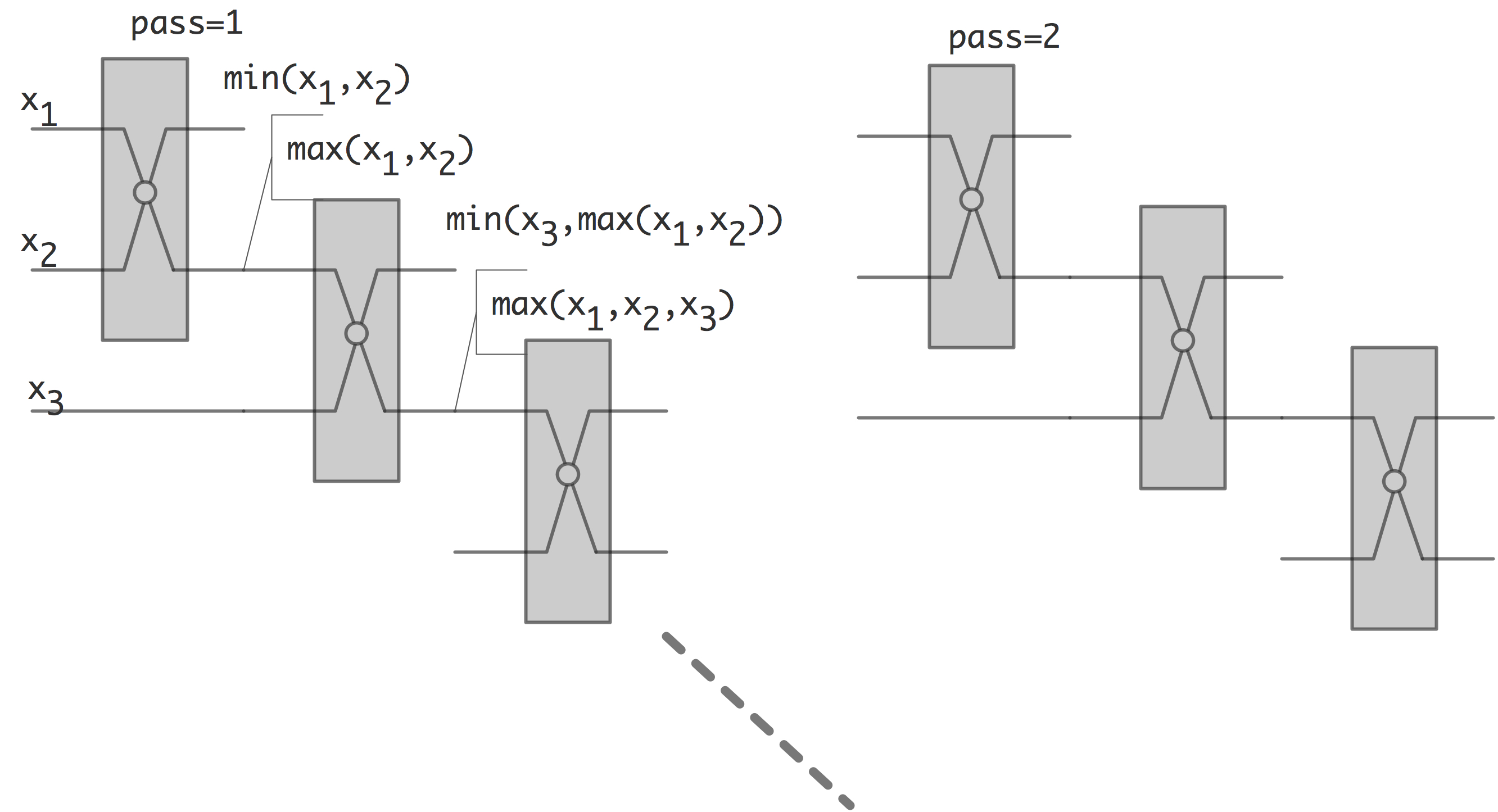
下面我们将讨论排序网络中的一个例子:双调排序。
并行复杂度
串行排序算法最理想的时间复杂度为$O(N \log N)$,理想情况下,如果同时使用$P=N$个处理器计算,我们希望并行排序的时间复杂度为$O(\log N)$。如果并行时间大于这个值,我们将所有处理器上的操作总数定义为串行复杂度(sequential complexity)。
这相当于如果并行算法由一个进程执行。理想情况下,这与单进程算法的操作数相同,但如果线程数过大,该算法就会收到并行算法的额外时间开销惩罚。
例如,下面我们将看到,排序算法的串行复杂度通常为$O(N \log_2 N)$。
奇偶交换排序
观察上图我们发现,pass 2实际上可以在pass 1完成前开始,在给定的时间下,这实际上就是奇偶交换排序(odd-even transposition sort)。
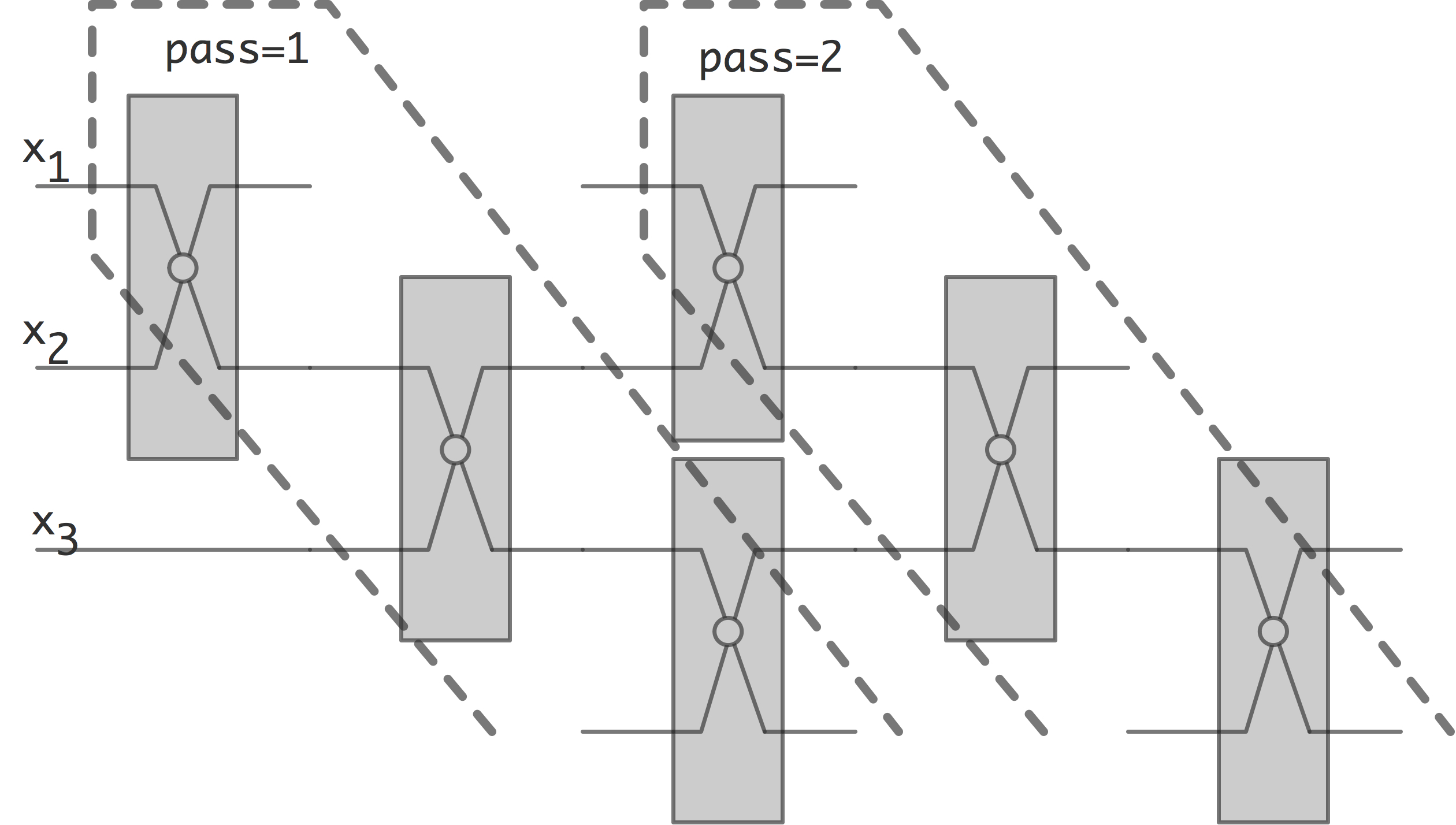
奇偶交换排序是一种简单的并行排序算法,其主要优点是在线性面积的处理器上容易实现;缺点是效率并不理想。
该算法的一个步骤由两个子步骤组成:
- 偶数的处理器与它的右邻进行比较和交换;然后
- 奇数处理器与它的右邻进行比较和交换。
定理 2 该算法经过$N/$步完成(其中每步由刚才给出的两个子步骤组成)。通过归纳法进行证明:在每个三联体$2i, 2i+1, 2i+2$中,经过一个偶数和一个奇数步骤后,最大的元素将处于最右的位置。
在并行时间为$N$的情况下,可以得到串行复杂度为$N^2$。
练习 8.1 讨论交换排序的速度和效率,我们对$N$个处理器的$P$个数字进行排序;设$N = P$,即每个处理器都包含一个数字。用比较和交换操作表示执行时间。
- 这个并行代码总共需要多少次比较和交换操作?
- 该算法需要多少个串行步骤?什么是$T_1, T_p,T_\infty, S_p, E_p$的$N$数排序?平均并行量是多少?
- 交换排序可以被认为是冒泡排序的并行实现。现在让$T_1$指的是(顺序的)冒泡排序的执行时间。这对$S_p$和$E_p$有何影响?
快速排序
快速排序是一种递归算法,与冒泡排序不同,它的复杂度并不确定。该算法由两部分组成,是基于序列的排列。
1 | Algorithm: Dutch National Flag ordering of an array |
我们无需证明,这可以在$O(n)$操作中完成。这样一来,快速排序就变成了
1 | Algorithm: Quicksort |
该算法的不确定性来源于基准元素的选择。最坏情况下,算法所选择的基准元素每次都是数组中唯一最小的元素,如此需要再次进行$n-1$次递归操作。不难看出,此时的运行时间为$O(n^2)$。另一方面,如果基准元素总是(接近)中位数,即大小适中的元素,那么递归调用的运行时间将大致相等,可以得到递归公式:
复杂度为$O(n \log n)$。现在考虑快速排序的并行实现。
在共享内存中进行快速排序
通过共享内存模型和递归调用的线程实现,可以将快速排序进行简单并行化,然而这种方法并不高效。
在一个长度为$n$的数组中,如果有理想的基准元素,在算法的最后阶段会有$n$个线程在活动。最理想的情况是,我们希望并行算法在$O(\log n)$的时间内运行,但是在这里,时间被第一个线程对数组的初始重新排序所支配。
练习 8.2 精确论证这个结论。这样并行化快速排序算法的总运行时间、速度提升和效率是多少?是否有办法使分割数组的效率更高?答案是肯定的,关键是要使用并行的前缀操作,见附录20。如果数组中的数值是$x_1,…,x_n$,我们使用并行前缀来计算有多少元素小于基准$\pi$。
有了这个方法,如果一个处理器看了$x_i$,并且$x_i$小于基准,那么它需要被移到数组中的$X_{i + 1}$的位置,那里的元素是根据基准分割的。同样地,我们可以知道有多少元素大于基准元素,并相应地移动这些元素。
这表明每一个基准步骤可以在$O(\log n)$的时间内完成,由于排序算法有$\log 𝑛$的步骤,整个算法的运行时间为$O((\log n)^2)$。快速排序算法的串行复杂度$(\log_2 N)^2$。
超立方体上的快速排序
从上一节可以看出,为了使快速排序算法有效地并行化,我们也需要使Dutch National Flag reordering并行化。让我们假设数组已经被划分到维度为$p$的超立方体的$d$个处理器上(意味着$p=2d$)。
在并行算法的第一步,我们选择一个基准元素,并将其广播给所有处理器。然后,所有处理器将在其本地数据上独立地应用重新排序。
为了将第一层中的红色和蓝色元素聚集在一起,现在每个处理器都与一个二进制地址相同的处理器配对,但最重要的一位除外。在每一对中,蓝色元素被送到在该位上有1值的处理器;红色元素被送到在该位上有0值的处理器。
经过这次交换(这是本地的,因此是完全并行的),地址为1xx的处理器拥有所有的红色元素,地址为0xx的处理器拥有所有的蓝色元素。前面的步骤现在可以在子库中重复进行。
这种算法在每一步都能保持所有的处理器工作;但是,如果选择的基准元素远离中位数,就容易出现负载不平衡的情况。此外,在排序过程中,这种负载不均衡也不会减少。
在一般的并行处理器上进行快速排序
快速排序也可以在任何具有处理器线性排序的并行机器上进行。我们首先假设每个处理器正好持有一个数组元素,而且,由于标志重排,排序将总是涉及一组连续的处理器。
一个数组(或递归调用中的子数组)的并行快速排序开始于在存储数组的处理器上构建一个二进制树。选择一个基准元素值并在树上广播。然后,树状结构被用来在每个处理器上计算左、右子树中有多少元素小于、等于或大于基准元素值。
有了这些信息,根处理器可以计算出红色/白色/蓝色区域将被储存在哪里。这个信息被下发到树上,每个子树都计算出其子树中元素的目标位置。
如果我们忽略了网络竞争,现在可以在单位时间内完成重新排序,因为每个处理器最多发送一个元素。这意味着每个阶段只需要花费时间来计算子树中蓝色和红色元素的数量,在顶层是$O(\log n)$,在下层是$O(\log n/2)$,依此类推。这使得速度几乎完美。
基数排序
大多数排序算法都是基于比较完整的项值。相比之下,基数排序(radix sort)在数字的位数上做一些部分排序阶段。每个数字值都有一个 “bin “被分配,数字被移入这些bin。将这些仓连接起来就得到了一个部分排序的数组,通过移动数字的位置,数组的排序会越来越多。
考虑一个最多两位数的例子,所以需要两个阶段。
重要的是,一个阶段的部分排序在下一个阶段得到了保留。归纳起来,我们最后会得到一个完全排序的数组。
并行基数排序
分布式内存排序算法已经对数据进行了明显的 “分档”,所以基数排序的并行实现是基于使用𝑃,即进程的数量作为基数。
我们用两个处理器上的例子来说明这一点,也就是说,我们看的是数值的二进制表示。

分析。
确定所考虑的数字,以及确定有多少本地值进入哪个仓都是本地操作。我们可以将其视为一个连接矩阵$C$,其中$C[i, j]$是进程$i$将发送至进程$j$的数据量。每个进程拥有其在该矩阵中的一行。
为了在洗牌中接收数据,每个进程都需要知道将从其他每个进程接收多少数据。这需要对连接矩阵进行 “转置”。在MPI术语中,这是一个全对全操作。MPI_Alltoall。
在这之后,实际的数据可以在另一个all-to-all操作中进行洗牌。然而,由于每个$i, j$组合的数量不同,我们需要MPI_Alltoallv程序。
有效小数的基数排序
完全可以让基数排序的阶段从最重要的数字到最小的数字进行,而不是相反。按顺序,这不会改变任何重要的东西。
然而
- 我们不是完全洗牌,而是在越来越小的子集里洗牌,所以即使是串行算法也会增加空间局部性(spatial locality)。
- 一个共享内存的并行版本将显示出类似的局部性改进。
- 一个MPI版本不再需要全对全的操作。如果我们认识到每一个下一阶段都将在一个子集的进程内,我们可以使用通信器分割机制。
练习 8.3 上面的位置性论证做得有点手忙脚乱。论证该算法可以以广度优先和深度优先的方式进行。讨论一下这种区别与位置性论证的关系。
样本排序
你在快速排序算法(第8.3节)中看到,有可能在排序算法中使用概率元素。我们可以将快速排序法中挑选一个基准的想法扩展为挑选有多少个处理器的基准元素。这不是对元素的一分为二,而是将元素分成与处理器数量一样多的 “桶”。然后,每个处理器对其元素进行完全并行的排序。
显然,如果不仔细选择桶的话,这种算法会出现严重的负载不均衡。随机抽取$p$元素可能还不够好;相反,需要对元素进行某种形式的抽样。相应地,这种算法被称为样本排序(Samplesort)[17]。
虽然桶排序一旦分配即为完全并行,但仍有一些问题:首先,采样是该算法的一个串行瓶颈。另外,将桶分配给处理器的步骤本质上是一个全对全的操作。
为了对此进行分析,假设有$P$个进程首先作为映射器,然后作为规约器发挥作用。让𝑁为数据点的数量,并定义一个块大小$b\equiv N/P$。该算法的处理步骤的成本是:
- 局部确定每个元素的bin,需要时间$O(b)$;以及
- 局部排序,我们可以假设其最佳复杂度为$b \log b$。
然而,洗牌步骤是不简单的。除非数据被部分预排序,否则我们可以预期洗牌是一个完全的全对全,其时间复杂度为$P\alpha+b\beta$。另外,这可能会成为一个网络瓶颈。请注意,在超立方体上的快速排序法中,从来没有出现过对导线的争夺。
练习 8.4 论证对于少量的进程,$P <<N$,这个算法有完美的加速,串行复杂度(见上文)为$N \log N$。
将这种算法与双调排序等排序网络相比较,这种排序算法看起来相当简单:它只有一个步骤的网络。前面的问题认为,在 “理想扩展”(工作可以增加,同时保持处理器数量不变)中,串行复杂性与串行算法相同。然而,在我们按比例增加功和处理器的弱缩放分析中,串行复杂度要差很多。
练习 8.5 考虑我们同时扩展$N、P$的情况,保持$b$不变。论证在这种情况下,洗牌步骤在算法中引入了一个$N^2$项。
通过MapReduce排序
terasort基准涉及在一个基于文件的大型数据集上进行排序。因此,它在某种程度上是大数据系统的一个标准。特别是,MapReduce是一个主要的候选者,见http://perspectives.mvdirona.com/2008/07/hadoop-wins-terasort/。
使用MapReduce,该算法的过程如下。
- 通过抽样或优先信息确定一组键值:键值是这样的,预计每对键值之间的记录数量大致相等。间隔的数量等于规约器进程的数量。
- 然后,映射器进程产生键/值对,其中键是区间或规约器编号。
然后,规约器进程执行局部排序。
我们看到,除了术语的变化之外,这实际上是sampleort。
双调排序
为了激励双调排序,假设一个序列$x = ⟨x_0, … … , x_{n - 1}⟩$由升序和降序组成。现在把这个序列分成两个等长的子序列,定义为:
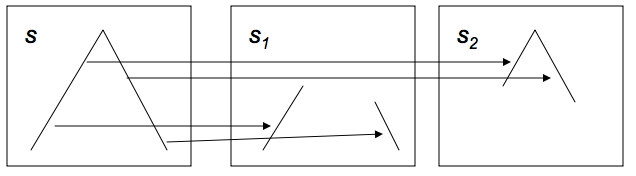
从图中不难看出,$s_1$, $s_2$又是具有升序和降序部分的序列。此外,$s_1$的所有元素都小于$s_2$的所有元素。
我们称(8.1)为升序双调拣器,因为第二个子序列中的元素比第一个子序列中的元素大。同样地,我们可以通过颠倒最大和最小的角色来构造一个降序分拣器。
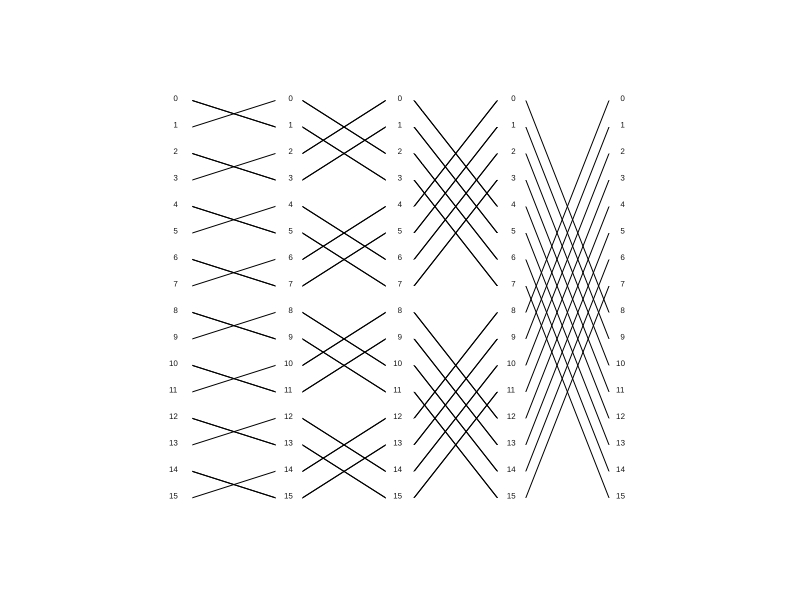
不难想象这是排序算法中的一个步骤:从这个形式的序列开始,递归应用公式(8.1)得到一个排序的序列。图8.4显示了4个位元排序器,分别在8,4,2,1的距离上,如何对一个长度为16的序列进行排序。
双调序列的实际定义要稍微复杂一些。如果一个序列由一个升序部分和一个降序部分组成,或者是这样一个序列的循环变异,那么它就是比特序列。
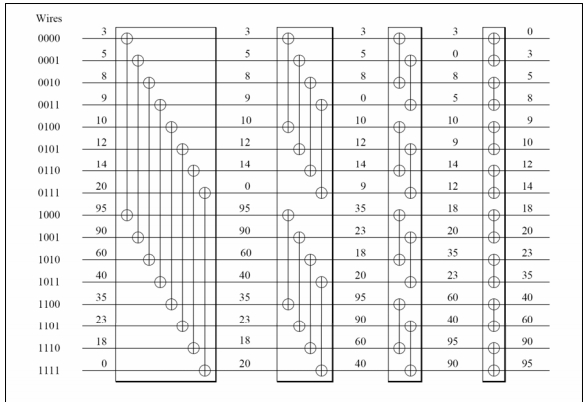
练习 8.6 证明根据公式(8.1)分割一个位数序列可以得到两个位数序列。
所以问题是如何得到一个比特尼序列。答案是使用越来越大的双调网络。两个元素的位数排序给你一个排序的序列。如果你有两个长度为2的序列,一个向上排序,另一个向下排序,这就是一个双调序列。因此,这个长度为四的序列可以通过两个比特级的步骤进行排序。
两个长度为四的排序形成一个长度为四的比特序列。可以用三个比特步骤进行排序;等等。
从这个描述中你可以看到,你要用$\log_2N$个阶段来排序$N$个元素,其中第$i$个阶段的长度是$\log_2i$。这使得位数排序的总顺序复杂性为$(\log_2 N )^2$。
图8.5中的操作序列被称为排序网络,由简单的比较-交换元素构成。元素组成的排序网络。与快速排序的情况不同,它不依赖于数据元素的值。
素数查找
埃拉托色尼筛选法是一种非常古老的寻找素数的方法。在某种程度上,它仍然是许多现代方法的基础。
写下从2到某个上限$N$的所有自然数,我们要把这些数字标记为素数或绝对非素数。所有的数字最初都是没有标记的。
- 第一个没有标记的数是2:把它标记为质数,并把它的所有倍数标记为非质数。
- 第一个没有标记的数字是3:把它标记为质数,并把它的所有倍数标记为非质数。
- 下一个数字是4,但它已经被标记过了,所以标记5和它的倍数。
- 下一个数字是6,但它已经被标记过了,所以标记7和它的倍数。
- 数字8,9,10已经被标记过了,所以继续标记11。
- 依此类推。
图论分析
科学计算中的各种问题都可以被表述为图问题(关于图论的介绍见附录18);例如,我们已经遇到了负载平衡(第2.10.4节)和寻找独立集(第6.8.2节)的问题。
许多传统的图算法不能立即适用,或者至少不能有效地适用,因为图往往是分布式的,而传统的图理论假设了对整个图的全局知识。此外,图论通常关注的是寻找最佳算法,而这通常不是一种并行的算法。因此,并行图算法本身就是一个研究领域。
最近,在科学计算中出现了新的图计算类型。这里的图不再是工具,而是研究对象本身。例如,万维网或Facebook的社交图,或一个生物体内所有可能的蛋白质相互作用的图。
由于这个原因,组合计算科学正在成为一门独立的学科。在这一节中,我们看一下图分析:大型图的计算。我们首先讨论一些经典的算法,但我们在代数框架中给出这些算法,这将使并行实现更加容易。
传统图算法
我们首先看一下几个 “经典 “的图算法,并讨论如何以并行方式实现它们。图和稀疏矩阵之间的联系(见附录18)在这里至关重要:许多图算法具有稀疏矩阵-向量乘法的结构。
最短路径算法
有几种类型的最短路径算法。例如,在单源最短路径算法中,人们想知道从一个给定节点到任何其他节点的最短路径。在全对最短路径算法中,人们想知道任何两个节点之间的距离。计算实际路径不是这些算法的一部分;但是,通常很容易包括一些信息,通过这些信息可以在以后重建路径。
我们从一个简单的算法开始:寻找非加权图中的单源最短路径。用广度优先搜索(BFS)来做这件事很简单。
1 | Input : A graph, and a starting node 𝑠 |
这种制定算法的方式在理论上是很有用的:通常可以制定一个谓词,在while循环的每一次迭代中都是真的。这样,你就可以证明该算法终止了,并且它计算了你想要计算的东西。在传统的处理器上,这的确是你对图算法的编程方式。然而,现在的图,如来自Facebook的图,可能是巨大的,而你想对你的图算法进行并行编程。
在这种情况下,传统的表述方式就显得不够了。
它们通常基于队列,节点被添加或减去;这意味着存在某种形式的共享内存。
关于节点和邻居的声明是在不知道这些是否满足任何形式的空间定位的情况下做出的;如果一个节点被触及一次以上,就不能保证时间上的定位。
Floyd-Warshall最短路径
Floyd-Warshall算法是一个全对最短路径算法的例子。它是一种动态编程算法,是基于逐步增加路径的中间节点集。具体来说,在步骤𝑘中,所有的路径$u\rightarrow v$都被考虑,它们的中间节点在集合 $k=\{0, …. \Delta k(u,v)\}$被定义为从$u$到$v$的路径长度,其中所有中间节点都在$k$。最初,这意味着只考虑图边,当$k \equiv |V|$时,我们已经考虑了所有可能的路径,算法完成。
计算的步骤是
也就是说,对距离$\Delta (u, v)$的第 $k$个估计值是旧的估计值和一条新的路径的最小值,现在我们正在考虑节点$k$的可行性。这条路径是由$u \rightarrow k$和$k \rightarrow v$两条路径连接而成的。
用算法来写:
1 | for 𝑘 from zero to |𝑉| do |
我们看到这个算法的结构与高斯消除法相似,只是在那里,内循环是 “对于所有的$u,v>k$”。
在第5.4.3节中,你看到稀疏矩阵的因式分解会导致填充,所以这里也会出现同样的问题。这需要灵活的数据结构,而且这个问题在并行时变得更加严重,见第9.5节。
代数上。
1 | for 𝑘 from zero to |𝑉| do |
Floyd-Warshall算法并不告诉你实际的路径。在上面的距离计算过程中,存储这些路径的成本很高,无论是时间还是内存。一个更简单的解决方案是可能的:我们存储第二个矩阵𝑛(𝑖, 𝑗),它有𝑖和𝑗之间路径的最高节点号。
练习 9.1 将$n(i, j)$的计算包含在 Floyd-Warshall 算法中,并描述在已知$d(⋅, ⋅)$和$n(⋅, ⋅)$的情况下,如何利用它来寻找$i$ 和$j$之间的最短路径。
生成树
在一个无向图$G=⟨V,E⟩$中,如果$T\subset E$是连接和无环的,我们称之为 “树”。如果它的边包含所有的顶点,则被称为生成树。如果图的边权重为$w_i ∶i\in E$,则树的权重为$\sum_{e\in T}w_e$,如果树的权重最小,我们称它是最小生成树。一个最小生成树不需要是唯一的。
Prim算法是Dijkstra最短路径算法的一个小变种,它从一个根开始计算生成树。根的路径长度为零,而所有其他节点的路径长度为无穷大。在每一步中,所有与已知树节点相连的节点都会被考虑,并更新它们的最佳已知路径长度。
1 | for all vertices 𝑣 do l(𝑣) ← ∞ |
定理3 上述算法计算出每个节点到根节点的最短距离。
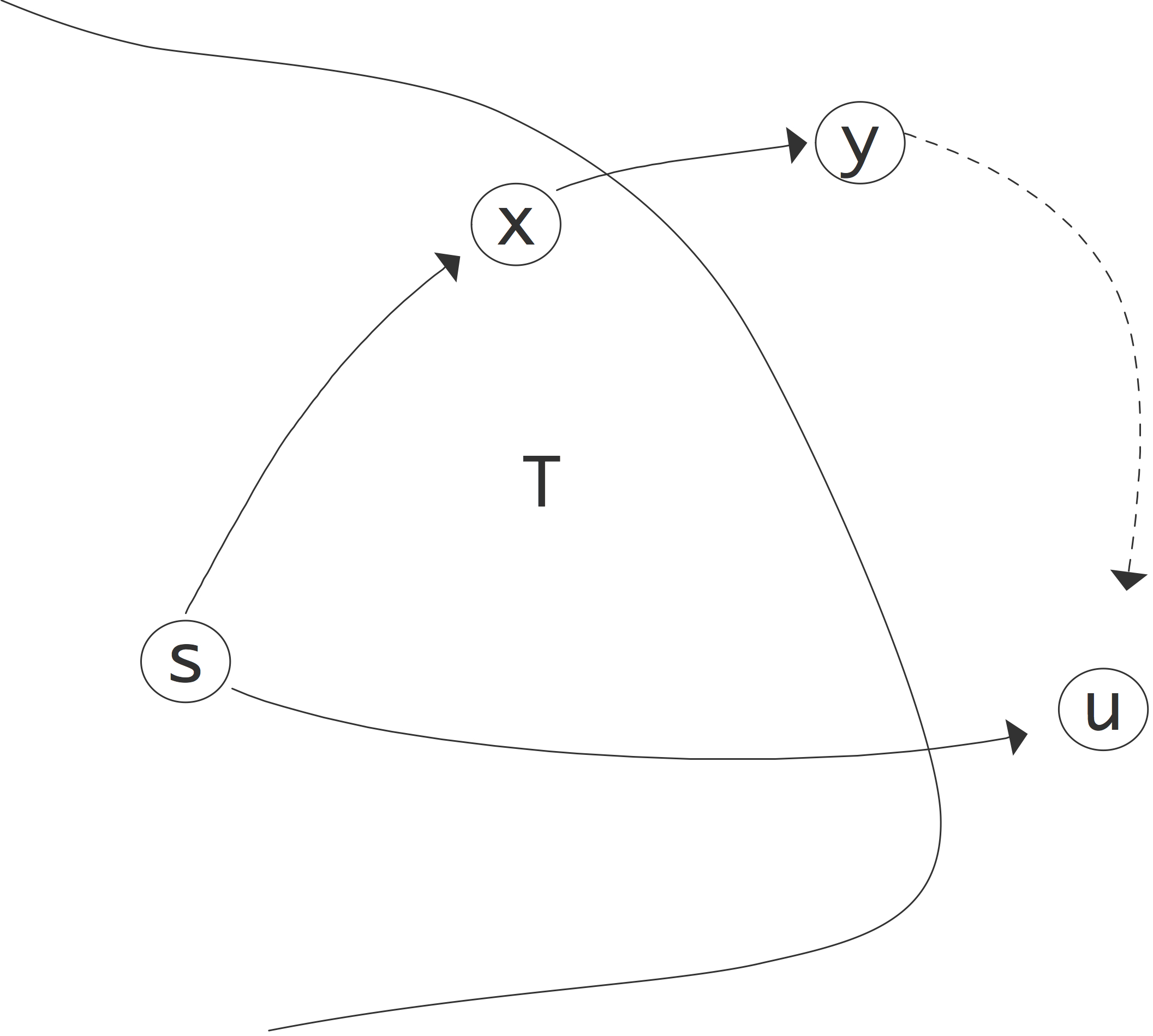
证明:本算法正确性的关键在于我们选择$u$为最小$l(u)$值。把到顶点的真正最短路径长度称为$L(v)$。由于我们从一个无限大的$l$值开始,并且只减少它,我们总是有$L(v)\leqslant l(v)$。
我们的归纳假设是,在算法的任何阶段,对于当前租用的$T$中的节点,路径长度的确定是正确的。
当树只由根$s$组成时,这显然成立。现在我们需要证明归纳步骤:如果对于当前树中的所有节点,路径长度都是正确的,那么我们也将得到$L(u)=l(u)$。假设这不是真的,有另一条路径更短。这条路径需要经过目前不在$T$中的某个节点$y$;这在图9.1中得到说明。假设$x$是$T$中的节点,位于$y$之前的据称最短路径。现在我们有$l(u)>L(u)$,因为我们还没有正确的路径长度,并且$L(u)>L(x)+w$,因为在$y$和$u$之间至少有一条边(有正权)。 但$x\in T$,所以$L(x)=l(x)$,$L(x)+y=l(x)+y$。现在我们注意到,当$x$被添加到$T$时,它的邻居被更新,所以$l(y)$是$lx+y$或更少。将这些不等式,我们发现
这与我们选择$u$为最小$l$值的事实相矛盾。
为了并行化这个算法,我们观察到内循环是独立的,因此可以并行化。然而,外循环有一个选择,即最小化一个函数值。计算这个选择是一个减法运算,随后它需要被广播。这种策略使得顺序时间等于$d \log P$,其中$d$是生成树的深度。
在单个处理器上,寻找数组中的最小值是一个$O(N)$的操作,但通过使用优先级队列,这可以减少到$O(\log N)$。对于生成树算法的并行版本,相应的项是$O(\log(N/P))$,还不包括减少的$O(\log P)$成本。
Graph cut
有时,你可能想对一个图进行分区,例如出于并行处理的目的。如果这是通过划分顶点来实现的,那么你就是在切割边,这就是为什么这被称为顶点切割分区。对于什么是好的顶点切割,有各种标准。例如,你希望切割的部分大小大致相等,以平衡平行工作。由于顶点通常对应于通信,你希望顶点的数量(或在加权图中它们的权重之和)要小。图Laplacian(第18.6.1节)是这方面的一个流行算法。
图的切割的另一个例子是二方图的情况:一个有两类节点的图,而且只有从一类到另一类的边。例如,这样的图可以模拟一个人口和一组属性:边表示一个人有某种兴趣。现在你可以做一个边的切割,将边的集合进行分割。这可以给你一些具有类似兴趣的人的集合。这个问题很有意思,比如说,用于定位在线广告。
并行化
许多图的算法,如第9.1节中的算法,并不容易并行化。这里有一些考虑。
首先,与许多其他算法不同,很难针对最外层的循环层,因为这通常是一个 “while “循环,使得并行停止测试成为必要。另一方面,通常有一些宏观步骤是连续的,但其中的一些变量是独立考虑的。因此,确实存在着可以利用的并行性。
图算法中的独立工作是一种有趣的结构。虽然我们可以确定 “for all “循环,它是并行化的候选者,但这些循环与我们以前看到的不同。
- 传统的公式往往具有逐步建立和删除的变量集的特征。这可以通过使用共享数据结构和任务队列来实现,但这限制了某种形式的共享内存的实现。
- 接下来,虽然每个迭代操作都是独立的,但其运行的动力学原理意味着将数据元素分配给处理器是很棘手的。固定分配可能会导致很多空闲时间,但动态分配会带来很大的开销。
- 在动态任务分配的情况下,算法将几乎没有空间或时间上的定位。
由于这些原因,线性代数的表述可能是比较好的。一旦考虑到分布式内存,我们肯定需要这种方法,但即使在多核架构上,它也能为鼓励局部性而付出代价。
在第6.5节中,我们讨论了稀疏矩阵-向量乘积的并行评估问题。由于稀疏性,只有按块行或块列进行划分才有意义。实际上,我们让分区由问题变量之一来决定。这也是唯一对单源最短路径算法有意义的策略。
练习 9.2 你能为将并行化建立在向量的分布上做一个先验的论证吗?这个操作涉及多少数据,多少工作,多少个顺序步骤?
策略
下面是三种图算法的并行化方法,按明显程度递减,按可扩展性递增。(另见[118])。
动态规划
许多图算法建立了一个要处理的顶点的数据结构$V$;他们执行一个包含循环的超步序列
1 | for all v in V: |
如果对一个顶点$v$的处理不影响$V$本身,那么这个循环是并行的,可以通过动态调度来执行。
这实际上是将数据动态分配给处理元素。在这个意义上,它是高效的,没有任何处理元素会与不需要处理的数据元素发生冲突,因此计算机的所有处理能力都得到了充分的利用。另一方面,动态分配带有操作系统的开销,而且会导致大量的数据传输,因为顶点不可能在处理元素的本地内存(如缓存)中。
另一个问题是,这个循环可能看起来像。
1 | for all v in V: |
现在可能发生的情况是,两个节点$v_1$和$v_2$都在更新一个共享的邻居$u$,这个冲突需要通过缓存一致性来解决。这带来了延迟上的损失,甚至可能需要使用锁,这带来了操作系统上的损失。
线性代数解释
现在我们将表明,图算法通常可以被视为稀疏矩阵算法,这意味着我们可以应用我们为这些算法开发的所有概念和分析。
如果$G$是图的邻接矩阵,我们也可以将最短路径算法类似于一系列矩阵-向量乘法(见附录18.5.4节)。让$x$是列出与源的距离的向量,也就是说,$x_i$是节点$i$与源的距离。对于$i$的任何邻居$j$,到源的距离是$x_i + G_{ij}$,除非已经知道更短的距离。换句话说,我们可以定义
而上述while循环的迭代对应于此定义下的后续矩阵-向量积。
这种算法之所以有效,是因为我们可以在第一次访问时将$d(v)$设置为其最终值:这恰恰发生在等于路径长度的外循环次数之后。内循环执行的总次数等于图中边的数量。加权图就比较麻烦了,因为一个有更多阶段的路径实际上可以用阶段的权重之和来衡量更短。下面是贝尔曼-福特算法。
1 | Let 𝑠 be given, and set 𝑑(𝑠) = 0 Set 𝑑(𝑣) = ∞ for all other nodes 𝑣 for |𝐸| − 1 times do |
这个算法是正确的,因为对于一个给定的节点$u$,在外迭代的$k$步之后,它已经考虑了所有路径$s\rightarrow u$的$k$阶段。
练习 9.3 这个算法的复杂度是多少?如果你对图形有一定的了解,外循环的长度是否可以减少?
我们可以再次将其写成一系列的矩阵-向量积,如果我们将积定义为
这与上述基础基本相同:到𝑗的最小距离是已经确定的距离的最小值,或者到任何节点𝑖的最小距离加上过渡$g_{ij}$。
全对算法的并行化
在上面的单源最短路径算法中,我们没有太多选择,只能通过分布向量而不是矩阵来实现并行化。这种类型的分布在这里也是可能的,它对应于$𝐷(⋅, ⋅)$量的一维分布。
练习 9.4 画出该算法变体的并行实现。证明每个$k$次迭代都涉及到以处理器$k$为根的广播。
然而,这种方法遇到了与使用一维矩阵分布的矩阵-向量乘积相同的缩放问题;见6.2.2节。因此,我们需要使用二维分布。
练习 9.5 做缩放分析。在内存不变的弱缩放情况下,渐近效率是多少?
练习 9.6 使用二维分布的$𝐷(⋅, ⋅)$量勾画Floyd-Warshall算法。
练习 9.7 并行的Floyd-Warshall算法对零点进行了相当多的操作,当然是在早期阶段。你能设计一种算法来避免这些操作吗?
分割
传统的图划分算法[146]并不是简单的可并行化。相反,有两种可能的方法。
- 使用图Laplacian;第18.6.1节。
- 使用多层次的方法。
1 分割图的粗化版本。
2 逐步解除图的粗化,调整分区。
- 使用多层次的方法。
现实世界 “图
在诸如第4.2.3节的讨论中,你已经看到了PDEs的离散化是如何导致具有图的方面的计算问题的。这种图的特性使它们适合于某些类型的问题。例如,使用FDMs或FEMs对二维或三维物体进行建模,会导致每个节点只与几个邻居相连的图形。这使得它很容易找到分离器,这反过来又允许像嵌套剖分这样的解决方法;见6.8.1节。
然而,有一些应用的计算密集型图问题看起来并不像FEM图。我们将简要地看一下全球网络的例子,以及像谷歌的PageRank这样试图寻找权威节点的算法。
现在,我们将称这种图为随机图,尽管这个术语也有技术含义[61]。
随机图的性质
我们在本课程的大部分内容中看到的图形,其属性源于它们是我们三维世界中物体的模型。因此,两个节点之间的典型距离通常是$O(N^{1/3})$,其中$N$是节点的数量。随机图的表现并非如此:它们通常具有小世界的特性,典型的距离是$O(\log N )$。一个著名的例子是电影演员和他们在同一电影中出现的联系图:根据 “六度分隔”,在这个图中没有两个演员的距离超过六。在图形方面,这意味着图形的直径是六。
小世界图还具有其他特性,例如存在小团体(尽管这些特性在高阶有限元问题中也有)和枢纽:高度的节点。这导致了如下的影响:在这样的图中删除一个随机的节点不会对最短路径产生很大的影响。
练习 9.8 考虑机场图和它们之间存在的航线。如果只有枢纽和非枢纽,论证删除一个非枢纽对其他机场之间的最短路径没有影响。另一方面,考虑到节点在二维网格中的排序,并删除一个任意的节点。有多少条最短路径会受到影响?
超文本算法
有几种基于线性代数的算法用于衡量网站的重要性[139]。我们将简要地定义几个算法并讨论其计算意义。
HITS
在HITS(Hypertext-Induced Text Search)算法中,网站有一个衡量它指向多少其他网站的枢纽得分,以及一个衡量有多少网站指向它的权威得分。为了计算这些分数,我们定义了一个发生率矩阵$L$,其中
权威分数$x$被定义为所有指向$i$的中心分数$y_j$的总和,反之亦然。因此
或$x=LL^tx$,$y=L^tLy$,表明这是一个特征值问题。我们需要的特征向量只有非负项;这被称为非负矩阵的Perron向量,见附录13.4。佩伦向量是通过幂方法计算的;见13.3节。
一个实用的搜索策略是。
- 找到所有包含搜索词的文档。
- 建立这些文档的子图,以及可能的与之相关的一到两层的文档。
- 计算这些文档的权威性和枢纽得分,并将其作为一个有序的列表呈现给用户。
PageRank
PageRank[166]的基本思想与HITS相似:它的模型是 “如果用户不断点击页面上某种程度上最理想的链接,那么总体上最理想的链接集合是什么”。这是通过定义一个网页的排名是所有连接到它的网页的排名之和来建模的。该算法 它通常是以迭代的方式表述。
1 | while Not converged do for all pages 𝑖 do |
其中的排名是这个算法的固定点。$\varepsilon$项解决了这样一个问题:如果一个页面没有出站链接,那么一个会在那里停留的用户就不会离开。
练习 9.9 论证这个算法可以用两种不同的方式来解释,大体上响应雅可比和高斯-赛德尔迭代法;5.5.3节。
练习 9.10 在PageRank算法中,每个页面都给它所连接的页面 “提供排名”。给出这个变体的伪代码。说明它对应于一个列的矩阵向量乘积,而不是上述表述的行。在共享内存并行中实现这个方法会有什么问题?
对于这个方法的分析,包括它是否收敛的问题,最好是完全用线性代数的术语来表述。我们再次定义一个连接矩阵
$e=(1,…,1)$,向量$d^t=e^tM$计算一个页面上有多少个链接。$d_i$是页面$i$上的链接数量。我们构建一个对角矩阵$D = diag(d_1, …)$ 我们将$M$归一为$T= MD^{-1}$。
现在$T$的列和(即任何一列的元素之和)都是1,我们可以表示为$e^t T = e^t$ 其中$e^t =(1,…,1)$。这样的矩阵被称为随机矩阵。它有如下解释。
如果$p$是一个概率向量,即$p_i$是用户正在查看页面$i$的概率,那么$T_p$是用户点击了一个随机链接后的概率向量。
练习 9.11 从数学上讲,概率向量的特点是其元素之和为1。说明随机矩阵与概率向量的乘积确实又是一个概率向量。
上面制定的PageRank算法对应于取一个任意的随机向量$p$,计算权力法$T_p,T^2_p,T^3_p,…,$并看这个序列是否收敛到什么。
这个基本算法有一些问题,例如没有外向链接的网页。一般来说,在数学上我们是在处理 “不变子空间”。例如,考虑一个只有2个页面的网络,并有以下邻接矩阵。
自己检查一下,这对应于第二页没有外链的情况。现在让$p$成为起始向量$p_t = (1, 1)$,并计算几个迭代的功率法。你是否看到,用户在第二页的概率上升到了1?这里的问题是,我们正在处理一个可还原矩阵。
为了防止这个问题,PageRank引入了另一个元素:有时候,用户会因为点击而感到无聊,会去一个任意的页面(对于没有出站链接的页面也有规定)。如果我们把𝑠称为用户点击一个链接的机会,那么进入一个任意页面的机会就是1-𝑠。一起来看,我们现在有这样一个过程
也就是说,如果$p$是一个概率向量,那么$p’$就是一个概率向量,它描述了用户在进行了一次页面转换(无论是通过点击一个链接还是通过 “传送”)之后的位置。
PageRank向量是这个过程的静止点;你可以把它看作是用户进行了无限次转换之后的概率分布。PageRank向量满足以下条件
因此,我们现在不得不想,$I-sT$是否有一个逆。如果逆的存在,它满足
不难看出逆的存在:利用格什哥林定理(附录13.5),你可以看到$T$的特征值满足$|\lambda| \leqslant 1$。现在利用$𝑠<1$,所以部分和的序列收敛。
上述逆的公式也指出了一种通过使用一系列矩阵-向量乘法来计算PageRank向量$p$的方法。
练习 9.12 写出计算PageRank向量的伪代码,给定矩阵$T$。证明你从来不需要明确计算$T$的幂。(这是霍纳规则的一个实例)。
在$𝑠=1$的情况下,也就是说,我们排除了远程传输,PageRank向量满足$p=Tp$,这又是寻找Perron向量的问题;见附录13.4。
我们通过幂级迭代找到佩伦向量(第13.3节)。
这是一个稀疏的矩阵向量乘积,但与BVP的情况不同,稀疏性不太可能有带状结构。在计算上,人们可能必须使用与密集矩阵相同的并行论据:矩阵必须是二维分布的[162]。
大型计算图论
在前面的章节中,你已经看到许多图算法有一个计算结构,使矩阵-向量积成为它们最重要的内核。由于大多数图相对于节点数来说是低度的,所以该乘积是一个稀疏的矩阵-向量乘积。
然后在很多情况下,我们可以像第6.5节那样,对矩阵进行一维分布,由图节点的分布诱导:如果一个处理器拥有一个图节点$i$,它就拥有所有的边$i$,$j$。
然而,往往计算是非常不平衡的。例如,在单源最短路径算法中,只有沿着前面的顶点是活跃的。由于这个原因,有时边的分布而不是顶点的分布是有意义的。为了平衡负载,甚至可以使用随机分布。
Floyd-Warshall算法的并行化(第9.1.2节)沿着不同的路线进行。在这里,我们不计算每个节点的数量,而是计算作为节点对的函数的数量,也就是一个类似矩阵的数量。因此,我们不是分配节点,而是分配节点对的距离。
节点与边的分布
有时我们在处理稀疏图矩阵时需要超越一维分解。我们假设我们要处理的图的结构或多或少是随机的,比如说,对于任何一对顶点来说,有一条边的机会是相同的。另外,假设我们有大量的顶点和边缘,那么每个处理器都会存储一定数量的顶点。那么结论是,任何一对处理器需要交换信息的机会都是一样的,所以信息的数量是$O(P)$。(另一种可视化的方法是看到非零点随机地分布在矩阵中)。这并不能提供一个可扩展的算法。
出路是把这个稀疏矩阵当作密集矩阵,并援引第6.2.2节的论据来决定矩阵的二维分布。(参见[207]中对BFS问题的应用;他们以图的形式制定了他们的算法,但是二维矩阵-向量乘积的结构是清晰可辨的)。二维乘积算法只需要处理器行和列的集合体,所以涉及的处理器数量为$O(\sqrt{P})$。
超稀疏数据结构
在稀疏矩阵的二维分布下,可以想象一些过程会出现行数为空的矩阵。从形式上看,如果非零的数量(渐进地)小于维度,那么一个矩阵被称为超稀疏。
在这种情况下,CRS格式可能是低效的,可以使用称为双压缩存储的东西[24]。
N体问题
在第4章中,我们研究了连续现象,例如在整个区间[0,1]中,受热棒在一定时间内的行为。也有一些应用,你可能对有限数量的点感兴趣。一个这样的应用是研究粒子的集合,可能是非常大的粒子,如行星或恒星,在重力或电力的影响下。(也可能有外力,我们将忽略这一点;我们还假设没有碰撞,否则我们需要纳入最近邻的相互作用)。这种类型的问题被称为N体问题;关于介绍,见http://www.scholarpedia.org/article/N-body_simulations(引力)。
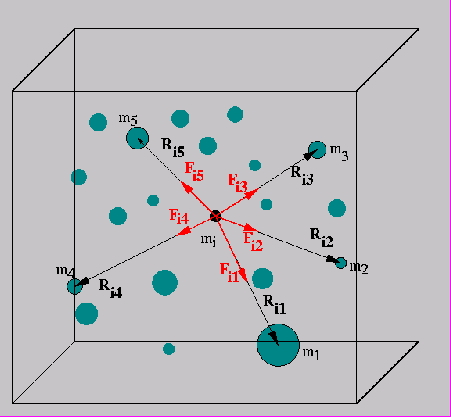
这个问题的基本算法是很容易的。
- 选择一些小的时间间隔。
- 根据所有粒子的位置,计算每个粒子上的力。
- 移动每个粒子的位置,就像它所受的力在整个时间间隔内保持不变一样。
对于一个足够小的时间间隔,这个算法给出了一个合理的近似事实。最后一步,更新粒子的位置,很容易而且完全并行:问题在于评估力。以一种天真的方式,这种计算足够简单,甚至是完全并行的。
1 | for each particle 𝑖 |
该算法的主要缺点为复杂度太高:对于$N$个粒子,操作的数量是 $O(N^2)$。
练习 10.1 如果我们有$N$个处理器,一个更新步骤的计算将需要时间$O(N)$。通信复杂度是多少?提示:是否可以用集合运算?
已经发明了几种算法来使顺序复杂度降低到$O(N \log N)$甚至是$O(N)$。正如可以预期的那样,这些方法比原始的算法更难实现。我们将讨论一种流行的方法:Barnes-Hut算法[8],其复杂度为$O(N \log N )$。
巴恩斯-胡特算法
导致复杂性降低的基本观察是以下几点。如果我们正在计算两个靠近的粒子$i_1,i_2$的力,这些力来自两个同样靠近的粒子$j_1,j_2$,你可以将$j_1,j_2$合并为一个粒子,并将其用于$i_1, i_2$。
接下来,该算法使用了空间的递归划分,在二维的象限和三维的八度空间中;见图10.2。
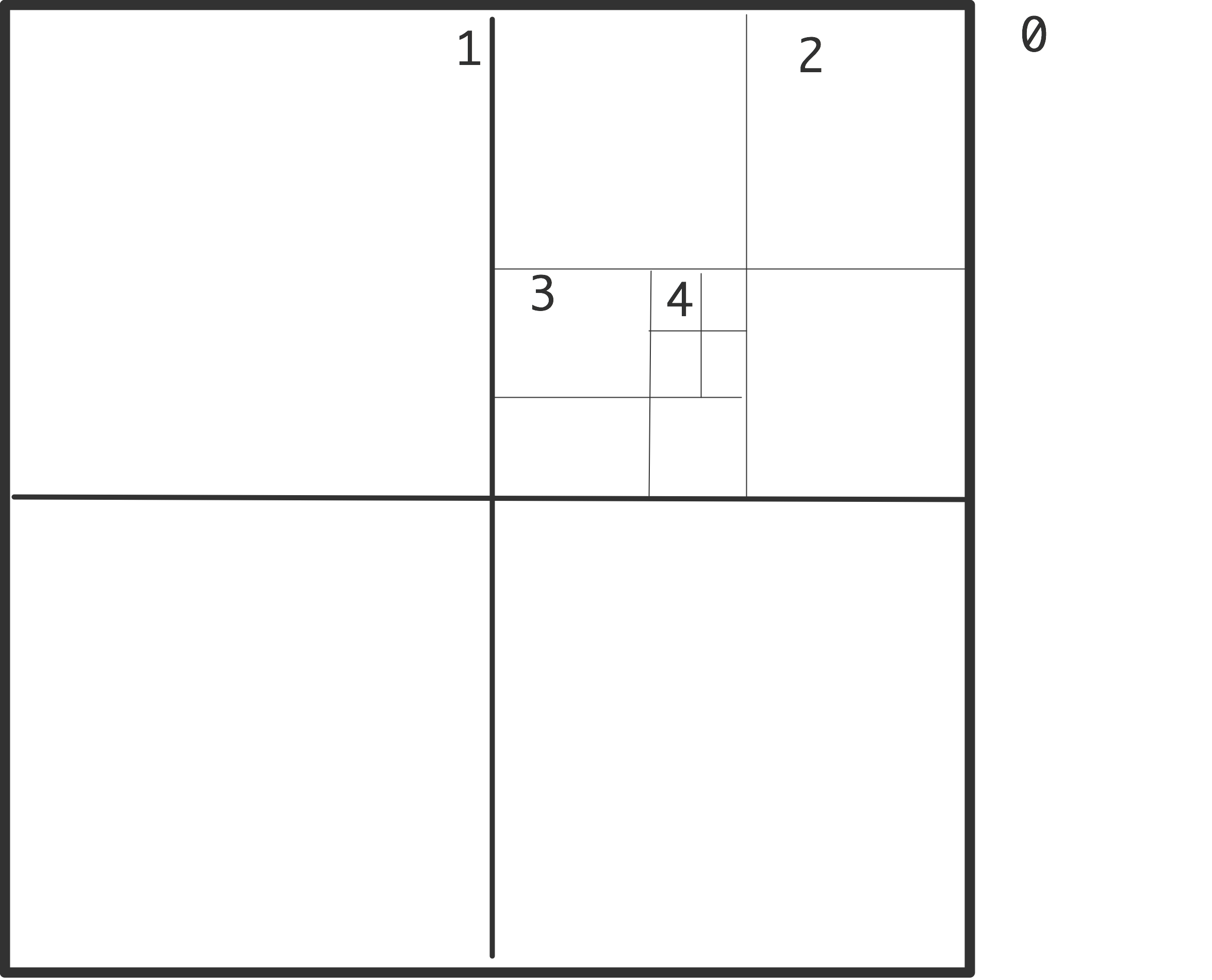
该算法如下。首先计算所有层的所有单元的总质量和质量中心。
1 | for each level l, from fine to coarse: for each cell 𝑐 on level l: |
这些级别被用来计算与每个粒子的相互作用。
1 | for each particle 𝑝: |
对一个细胞是否足够远的测试通常是以其直径与距离足够小的比率来实施的。这有时被称为 “细胞开放标准”。以这种方式,每个粒子与若干同心环的细胞相互作用,每个下一个环的宽度是双倍的;见图10.3。
如果细胞被组织成一棵树,这种算法就很容易实现。在三维的情况下,每个单元有八个孩子,所以这被称为八叉树。
质量中心的计算必须在粒子移动后每次进行。更新可能比从头开始计算的成本要低。另外,可能会发生一个粒子穿过一个单元的边界,在这种情况下,数据结构需要被更新。在最坏的情况下,一个粒子移动到一个原来是空的单元。
快速多极点法
快速多极法(FMM)计算每一点上的势的表达式,而不是像巴恩斯-胡特那样计算力。FMM使用的信息比盒子里的粒子的质量和中心还要多。这种更复杂的扩展更准确,但也更昂贵。作为补偿,FMM使用一组固定的盒子来计算势,而不是一组随精度参数θ和质心位置变化的盒子。
然而,在计算上,FMM很像Barnes-Hut方法,所以我们将共同讨论它们的实施。
全程计算
尽管有上述明智的近似方法,但也有对$N^2$相互作用进行全面计算的努力;例如,见Sverre Aarseth的NBODY6代码;见http://www.ast.cam.ac.uk/~sverre/web/pages/home.htm。这些代码使用高阶积分器和自适应时间步长。在Grape计算机上的快速实现是存在的;由于需要定期的负载平衡,一般的并行化通常是困难的。
执行
八叉树方法在高性能架构上提供了一些挑战。首先,问题是不规则的,其次,不规则性是动态变化的。第二方面主要是分布式内存中的问题,它需要负载再平衡;见2.10节。在本节中,我们集中讨论了单步的力计算。
矢量化
如图10.2的问题结构是很不规则的。这对于小规模的SSE/AVX指令和大规模的矢量流水线处理器的矢量化来说都是一个问题(关于两者的解释见2.3.1节)。程序步骤 “为某一盒子的所有孩子做某事 “将是不规则的长度,而且数据可能不会以规则的方式存储。
这个问题可以通过细分网格来缓解,即使这意味着有空盒子。如果底层被完全划分,将始终有八个(三维)粒子可以操作。更高的层次也可以被填满,但这意味着低层的空盒子数量越来越多,所以在增加工作和提高效率之间要进行权衡。
共享内存的实现
在顺序结构上执行,这个算法的复杂度为$O(N \log N)$。很明显,如果每个粒子变成一个任务,这个算法在共享内存上也可以工作。由于不是所有的单元都包含粒子,任务将有不同的运行时间。
分布式内存的实现
上述Barnes-Hut算法的共享内存版本不能立即用于分布式内存环境,因为每个粒子原则上可以从总数据的任何部分获取信息。使用哈希八叉树有可能实现这种思路的实现,但我们不会坚持这样做。
我们观察到,数据访问的结构化程度比最初看起来要高。考虑一个粒子$p$和它所交互的第l层的单元。位于𝑝附近的粒子将与相同的细胞互动,因此我们可以通过查看第l层的细胞和与之互动的同一层的其他细胞来重新安排互动。
这给我们提供了以下算法[123]:质心的计算变成了计算粒子$𝑔(l)$对第$l$层施加的力。
1 | for level l from one above the finest to the coarsest: |
有了这个,我们就可以计算出一个细胞上的力。
1 | for level l from one below the coarses to the finest: for each cell 𝑐 on level l: |
我们看到,在每一层,每个细胞现在只与该层的少量邻居互动。在算法的前半部分,我们只使用单元格之间的父子关系来向上攀登。据推测这是很容易的。
算法的后半部分使用更复杂的数据访问。第二项中的单元格𝑖都与我们要计算力的单元格𝑐有一定距离。在图的术语中,这些单元可以被描述为表亲:𝑐的父母的兄弟姐妹的孩子。如果开放的标准更加明确,我们就用二表兄妹:$c$的祖父母的兄弟姐妹的孙子,等等。
练习 10.2 论证这个力计算操作在结构上与稀疏矩阵-向量乘积有很多共同之处。
在共享内存的情况下,我们已经说过,不同的子树需要不同的时间来处理,但是,由于我们可能有更多的任务,而不是处理器核心,这一切都会扯平。对于分布式内存,我们缺乏将工作分配给任意处理器的可能性,所以我们需要谨慎地分配负载。空间填充曲线(SFC)可以在这里得到很好的应用(见2.10.5.2节)。
完整方法的1.5D实施
有可能通过分布粒子,让每个粒子评估来自其他每一个粒子的力,来直接实现完整的𝑁2方法的并行化。
练习 10.3 由于力是对称的,我们可以通过让粒子𝑝𝑖只与粒子𝑝𝑗 > 𝑖相互作用来节省2倍的工作量。在这个方案中,数据(如粒子)的平等分布会导致工作(如相互作用)的不均衡分布。粒子需要如何分布才能得到均匀的负荷分布?
假设我们不关心使用力的对称性所带来的两个系数的收益,而使用粒子的均匀分布,这个方案有一个更严重的问题,即它渐进地没有规模。
这方面的通信成本是
- $O(P)$延迟,因为需要从所有处理器接收信息。
- $O(N)$带宽,因为所有粒子都需要被接收。
然而,如果我们分配力的计算,而不是粒子,我们就会得出不同的界限;见[54]和其中引用的参考文献。
随着每个处理器计算边长为$N/\sqrt{P}$的区块中的相互作用,存在粒子的复制,需要收集力。因此,现在的成本是
- $O(\log p)$广播和还原的延迟
- $O(N/\sqrt{P}⋅\log P)$带宽,因此,每个处理器对最终的总和贡献了多少力。
问题描述
我们对N体问题进行如下抽象。
- 我们假设有一个粒子电荷/质量的向量$c(⋅)$和位置信息。
- 为了计算位置更新,我们需要基于存储图元$C_{ij}=
- 然后,这些相互作用被汇总为一个力矢量$f(⋅)$。
在并行性综合模型(IMP)模型中,如果我们知道各自的数据分布,就可以充分地描述算法;我们并不立即关注局部计算的情况。
颗粒分布
基于粒子分布的实现以分布为𝑐的粒子向量$c(u)$为起点,并直接计算同一分布上的力向量$f(u)$。我们将计算描述为三个内核的序列,两个是数据移动,一个是局部计算。
最后的内核是一个具有相同的$\alpha$和$\beta$分布的还原,所以它不涉及数据运动。本地计算内核也没有数据运动。这就留下了从初始分布$u(u)$收集$C(u,∗)$的问题。如果没有任何关于分布$u$的进一步信息,这就是对$N$元素的全收集,其成本为$\alpha \log p+\beta N$。特别是,通信成本不会随着$p$的下降而下降。
通过一个合适的基于分布的编程系统,方程组(10.1)可以被转化为代码。
工作分布
粒子分布的实现使用了一个一维分布$F(u,∗)$,用于与粒子分布一致的力。由于$F$是一个二维物体,所以也可以使用二维分布。我们现在将探讨这个选项,它早先在[54]中描述过;我们在这里的目的是要说明如何在IMP框架中实现和分析这个策略。我们不是把算法表达为分布式计算和复制的数据,而是使用分布式的临时,我们把复制方案推导为子通信器上的集体。
对于二维分布的$F$,我们需要$(N/b)\times (N/b)$ 处理器,其中$b$是一个块大小参数。为了便于说明,我们使用$b=1$,给出处理器的数量$P=N^2$。
现在我们将进行必要的推理。图10.4中给出了接近可实现形式的完整算法。为了简单起见,我们使用识别分布$I ∶p \rightarrow \{p\}$。
初始存储在处理器网格的对角线上 我们首先考虑收集图元 $C_{ij}=
为了正式表达这一点,我们让$D$为对角线$\{
力计算的复制 本地力计算$f_{pq} = f(pq)$对每个处理器$
以至于
(其中$p$代表将每个处理器映射到索引值$p$的分布,$q$ 同理)。
为了找到从$\alpha$到$\beta$分布的转化,我们考虑表达式$C$的转化$(D: )$。一般来说,任何集合$D$都可以写成从第一个坐标到第二个坐标的数值集合的映射。
在我们对角线的特殊情况下,我们有
有了这个,我们就可以写出
请注意,方程(10.4)仍然是$\alpha$分布。$\beta$分布是 𝐶(𝐼,𝑝),这是一个模式匹配的练习,可以看到这是由每一行的广播来实现的,其成本为$\alpha \log \sqrt{p}+\beta N/\sqrt{P}$。
同样地,$C(q,I)$是通过列广播从$\alpha$分布中找到的。我们的结论是,这个变体确实有一个通信成本,并随着处理器数量的增加而成比例地下降。
局部交互计算 $F(I , I)\leftarrow C(I, I)$具有相同的$\alpha$和$\beta$分布,因此它是并行的。
力的求和 我们必须将力矢量$f(⋅)$扩展到我们的二维处理器网格,即$f(⋅, ⋅)$。然而,为了符合对角线上的初始粒子分布,我们只在对角线上求和。该指令
有一个$\beta$ -分布的$I,D∶ ∗$ ,它是由$\alpha$ -分布的$I$,$I$通过聚集在行中形成。
蒙特卡洛方法
蒙特卡洛模拟是一个广义概念,指的是使用随机数和统计抽样来解决问题的方法,而非精确建模。从这种抽样的性质来看,结果会有一些不确定性,但统计学上的 “大数定理 “会确保不确定性随着样本数量的增加而下降。
统计抽样的一个重要工具是随机数发生器。参见教程32,了解随机数生成。
激励
在边长为2的正方形中随机选取一个点,它落入正方形内切圆中的概率为$\pi/4$。因此我们可以通过大量随机选取点来估计$\pi$的值。我们甚至可以做一个简单的物理实验:假设院子中存在一个形状不规则的池塘,而院子本身是面积已知的长方形。如果现在向院子里扔卵石,使它们在任何给定的地方都有同样的可能性,那么落在池塘里的卵石和落在外面的卵石的比例就等于面积的比例。
从数学角度讲,令$\Omega\in [0,1]^2$为某个区域,函数$f(\tilde{x})$为$\Omega$的边界,即
现在取随机点$\tilde{x}_0,\tilde{x}_1,\tilde{x}_2在[0,1]^2$,我们可以通过计算$f(\bar{x}_i)$为正或负的频率来估计Ω的面积。
我们可以把这个想法扩展到积分。一个函数在一个区间$(a, b)$上的平均值定义为
另一方面,我们也可以估计其平均数为
如果点$x$的分布合理,并且函数$f$不是太苛刻的话。可以得出
统计理论告诉我们,积分中的不确定性$\sigma_I$与下列因素有关。标准差$\sigma_f$的表达为
且服从正态分布。
吸引人的地方是什么?
到目前为止,蒙特卡洛积分看起来与经典的黎曼和积分没有什么区别,但当增加空间维度时,差别就较为明显。此时,经典积分需要在每个维度上有$N$个点,因此有$d$维上有$N^d$个点。另一方面,在蒙特卡洛方法中,点是从$d$维空间中随机抽取的,而且点的数量少得多。
在计算方面,蒙特卡洛方法很有吸引力,因为所有的函数评估都可以并行进行。
对标准差为$\sigma$的事件进行$N$次取样观察,其平均标准差为:$\sigma / \sqrt{N}$。这意味着更多采样观测将降低平均标准差以提高精度;使蒙特卡洛方法的有趣之处在于,这种准确性的提高与原始问题的维度无关。
蒙特卡洛方法是模拟具有统计性质现象的不二选择,如放射性衰变或布朗运动。而蒙特卡洛方法其他应用范畴则不属于科学计算领域,例如,用于股票期权定价的Black-Scholes模型[15]就使用了蒙特卡洛模拟。
线性方程组也可以通过蒙特卡洛方法求解,但这并不常见。下面我们将以一个用精确方法解答十分复杂的应用为例,说明蒙特卡罗方法所代表的统计抽样手段的便捷之处。
示例
伊辛模型的蒙特卡洛模拟
伊辛模型(介绍见[33])最初是为了模拟铁磁性而提出的。磁性是原子排列其 “自旋 “方向的结果:假设自旋只能是 “向上 “或 “向下”,那么如果有更多的原子自旋向上,而不是向下,那么这种材料就具有磁性,反之亦然。这些原子被称为 “晶格 “结构中的原子。
现在想象一下,加热一种材料会使原子松动。如果在材料上施加一个外部场,原子将开始与场对齐,如果场被移除,磁性又会消失。然而在某个临界温度以下,材料将保留其磁性。我们将使用蒙特卡洛模拟来寻找保留的稳定构型。
假设晶格$\Lambda$有$N$个原子,我们把原子的构型表示为$\sigma=(\sigma_1,…,\sigma_N)$,其中每个$\sigma_i=\pm1$。 晶格的能量模型为
第一个项模拟单个自旋$\sigma_i$与强度为$J$的外部场的相互作用。第二项是对近邻对的求和,模拟原子对的排列:如果原子具有相同的自旋,则乘积$\sigma_i\sigma_j$为正,如果相反则为负。
在统计力学中,构型的概率是
其中 “分区函数 “$Z$定义为
其中,总和在所有$2^N$配置上运行。
如果一个构型的能量在微小扰动下没有减少,那么它就是稳定的。为了探索这一点,我们在晶格上进行迭代,探索改变原子的自旋是否会降低能量。我们引入了一个偶然因素来防止人为的解决方案。(这就是Metropolis算法[155])。
1 | for fixed number of iterations do for each atom 𝑖 do |
如果我们注意到与稀疏矩阵-向量乘积结构的相似性,这种算法可以被并行化。在该算法中,我们也是通过结合几个近邻的输入来计算一个局部数量。这意味着我们可以对晶格进行分区,在每个处理器收集到一个幽灵区域(ghost region)后计算局部更新。
让每个处理器在晶格中的局部点上迭代,相当于晶格的一个特定的全局排序;为了使并行计算等同于串行计算,我们还需要一个并行随机发生器(第32.3节)。
机器学习
机器学习(Machine Learning,ML)是一些处理 “智能 “问题的技术的统称,如图像识别。抽象地讲,这些问题是从一个特征向量(features)空间(如图像中的像素值)到另一个结果向量空间的映射。在图像识别字母的情况下,这个最终空间可能是26维的,第二个分量的最大值将表明一个 “B “被识别。
ML技术的基本特征是,这种映射是由大量的内部参数描述的,而且这些参数是逐步完善的。这里的学习方面是,通过将输入与基于当前参数的预测输出和预期输出进行比较来完善参数。
神经网络
现在最流行的ML形式是深度学习(DL),或神经网络(neural networks)。神经网络,或深度学习网络,是一个计算数字输出的函数,给定一个(通常是多维的)输入点。假设这个输出被归一化为区间[0, 1],我们可以通过在输出上引入一个阈值来使用神经网络作为一个分类工具。
为什么是 “神经的”网络?
在生物体内,神经元是一个 “发射 “的细胞,也就是说,如果它收到某些输入,就会发出一个高电压。在ML中,我们把它抽象为一个感知器:一个根据某些输入而输出数值的函数。具体来说,输出值通常是输入的线性函数,其结果限制在(0,1)范围内。
单个数据点
在其最简单的形式中,我们有一个输入,其特征是一个特征向量$s\bar{x}$,和一个标量输出$y$。我们可以通过使用相同大小的权重向量和标量偏置𝑏来计算$y$作为$x$的线性函数。
激活函数
为了有一定的尺度不变性,我们引入了一个被称为激活函数的函数$\sigma$,它映射了$\mathbb{R}\rightarrow (0,1)$,我们实际上计算了标量输出$y$为
一种常见的sigmoid函数为
该函数有一个有趣的特性,即
所以计算函数值和导数并不比只计算函数值开销大多少。
对于向量值的输出,我们以点的方式应用sigmoid函数。
1 | // funcs.cpp |
其他激活函数有:$y=tanh(𝑥)$或’ReLU’(矫正线性单元)。
在其他地方(如DL网络的最后一层),softmax函数可能更合适。
多维输出
由$\bar{w}$、$\bar{b}$定义的单层可以实现我们对神经网的所有要求,这一点很罕见。通常情况下,我们使用一个层的输出作为下一个层的输入。这意味着我们计算的不是一个标量$y$,而是一个多维向量$\bar{y}$。
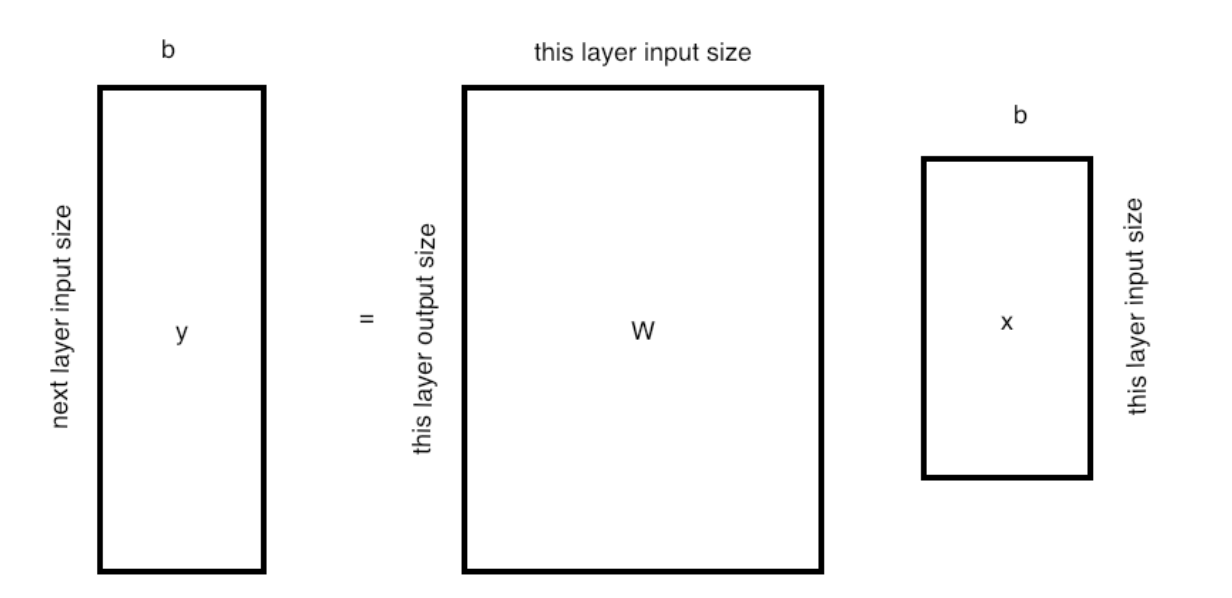
现在我们对输出的每个分量都有权重和偏置,所以
其中$W$现在是一个矩阵。
有几点看法:
如上所述,输出向量通常比输入的分量少,所以矩阵不是正方形,特别是不能倒置。
sigmoid函数使整个映射变得非线性。
神经网络通常有多个层次,每个层次都是以$x \rightarrow y$ 的形式进行的映射。
以上。
卷积
上面关于应用权重的讨论将输入视为一组没有进一步结构的特征。然而,在图像识别等应用中,输入向量是一个图像,有一个结构需要被承认。对输入向量进行线性化后,如果输入向量中的像素在水平方向上很接近,但在垂直方向上则不接近。
因此,我们的动机是找到一个能反映这种位置性的权重矩阵。我们通过引入内核来做到这一点:一个小的 “stencil”被应用于图像的各个点。(见第4.2.4节关于PDEs背景下的stencil的讨论)这样的内核通常是一个小的正方形矩阵,应用它是通过取模版值和图像值的内积来完成的。(这是对信号处理中卷积一词的不精确使用)。
例如:https://aishack.in/tutorials/image-convolution-examples/。
深度学习网络
现在我们将介绍一个完整的神经网络。这种介绍遵循[105]。
使用一个具有$L\geqslant 1$层的网络,其中 $\ell = 1$层是输入层,$\ell = L$层是输出层。
对于$\ell = 1,…,L$,第$\ell$层计算
其中,$a^{(1)}$是输入,$z^{(L+1)}$是最终输出。我们将其简洁地写成
其中我们通常会省略网对$W^{(\ell)}$、$𝑏^{(\ell)}$集的依赖。
1 | // net.cpp |
分类
在上述描述中,输入$x$和输出$y$都是向量值。也有一些情况是需要不同类型的输出的。例如,假设我们想描述数字的位图图像;在这种情况下,输出应该是一个整数0⋯9。
我们通过让输出$y$在$\mathbb{R}^{10}$中来解决这个问题,如果$y_5$比其他输出成分足够大,我们就说网络能识别数字5。通过这种方式,我们保持整个故事仍然是实值的。
误差最小化
通常我们有数据点${x_i}_{i=1,𝑁},$有已知的输出$y_i$,我们想让网络预测尽可能好地再现这种映射。从形式上看,我们寻求最小化成本,或者说误差。
在所有选择${W}$,${b}$中。(通常我们不会明确说明这个成本是所有$W^{[\ell]}$权重矩阵和$b^{[\ell]}$偏差的函数。)
1 | float Net::calculateLoss(Dataset &testSplit) { |
成本最小化意味着选择权重$\{W^{(\ell)}\}_\ell$和偏置$\{b^{(\ell)}\}_\ell$,使得对于每个$x$。
其中$L(N(x),y)$是一个损失函数,描述计算出的输出$N(x)$与预期输出$y$之间的距离。
我们使用梯度下降法找到这个最小值。
其中
这是一个复杂的表达式,我们现在将不做推导。
系数的计算
我们对成本相对于各种权重、偏差和计算量的部分导数感兴趣。对于这一点,引入一个简短的方法是很方便的。
现在应用连锁法则(完整的推导见上面引用的论文),我们得到,用$x \circ y$表示点式(或哈达玛)向量-向量积$\{x_iy_i\}$。
在最后层
递归到前一层级
对偏置量的敏感性
对权重的敏感性
使用特殊形式
给出

算法
现在我们在图12.1中介绍完整的算法。我们的网络有$\ell = 1, … , L$,其中参数$n_\ell$表示层$\ell$的输入大小。
第1层有输入$x$,第$L$层有输出$y$。考虑到minibatch的使用,我们让$x$,$y$表示一组大小为$b$的输入/输出,因此它们的大小分别为$n_1\times b$和$n_{L+1}\times b$。
计算层面
在本节中,我们将讨论深度学习的高性能计算方面。在标量意义上,我们论证了矩阵-矩阵乘积的存在,它可以被高效率地执行。我们还将讨论并行性,重点是数据并行,其基本策略是分割数据集。
- 模型并行,其基本策略是分割模型参数;以及
- 流水线化,在这种情况下,指令可以按照比原始的模型更多的串行执行。
重量矩阵乘积
无论是在应用网,即前向传递,还是在学习,即后向传递中,我们都要对权重矩阵进行矩阵乘向量的乘积。这种操作没有太多的缓存重用,因此性能不高;1.7.11节。
另一方面,如果我们将一些数据点捆绑在一起—这有时被称为小批量—并对它们进行联合操作,我们的基本操作就变成了矩阵乘以矩阵乘积,它能够有更高的性能;1.6.1.2节。
我们在图12.2中描述了这一点。
输入批和输出批由相同数量的向量组成。
权重矩阵$W$的行数等于输出大小,列数等于输入大小。相当于输入规模。
gemm内核的这种重要性(1.6.1和6.4.1节)导致人们为其开发专用硬件。
另一种方法是使用权重矩阵的特殊形式。在[143]中,研究了用托普利茨矩阵的近似。这在节省空间方面有优势,而且可以通过FFT进行乘积。
权重矩阵乘积的并行性
我们现在可以考虑有效计算$N(x)$。前面已经说过,矩阵-矩阵多运算是一个重要的内核,但除此之外,我们还可以使用并行处理。图12.3给出了两种并行化策略。
在第一种策略中,批处理被划分到各个进程中(或者说,多个进程同时处理独立的批处理);我们把这称为数据并行。
练习 12.1 考虑在共享内存环境下的这种情况。在这段代码中。
1 | for 𝑏 = 1, ... , batchsize |
假设每个线程计算1,…的部分内容。, batchsize的范围。把这一点翻译成你最喜欢的编程语言。你是按行还是列来存储输入/输出向量?为什么?这两种选择的影响是什么?
练习 12.2 现在考虑分布式内存背景下的数据并行,每个进程都在批处理的一个片断(块列)上工作。你看到一个直接的问题了吗?
还有第二种策略,被称为模型并行,其中模型参数,也就是权重和偏差是分布式的。正如在图12.3中看到的,这立即意味着该层的输出向量是分布式计算的。
练习 12.3 概述一下分布式内存中第二个分区的算法。
这些策略的选择取决于模型是否很大,权重矩阵是否需要被分割,或者输入的数量是否很大。当然,也可以把这些结合起来,模型和批处理都是分布式的。
权重更新
权重更新的计算
是一个等级为$b$的外积。它需要两个向量,并从它们中计算出一个低秩矩阵。
练习 12.4 根据$n_\ell$和$b$之间的关系,讨论该操作中(潜在)的数据重用量。为简单起见,假设$n_{\ell+1}\approx n_\ell$。
练习 12.5 讨论图 12.3 的两种分区策略中所涉及的数据移动结构。
除了这些方面,当我们考虑并行处理小批量时,这个操作变得更加有趣。在这种情况下,每个批次都独立地计算一个更新,我们需要对它们进行平均。在假设每个进程计算一个完整的$\Delta W$的情况下,这就变成了一个全还原。这种 “HPC技术 “的应用被开发成Horovod软件[76, 181, 111]。在一个例子中,在涉及40个GPU的配置上显示了相当大的速度。
另一个选择是延迟更新,或以异步方式执行。
练习 12.6 讨论在MPI中实现延迟或异步更新的问题。
流水线
最后一种类型的并行可以通过在各层上应用流水线来实现。简要说明这如何能提高训练阶段的效率。
卷积
应用卷积相当于乘以一个托普利茨矩阵。这比完全通用的矩阵-矩阵乘法的复杂度要低。
稀疏矩阵
权重矩阵可以通过忽略小条目而被稀疏化。这使得稀疏矩阵乘以密集矩阵的乘积成为主要的操作[72]。
硬件支持
从上述内容中,我们可以得出结论,gemm计算内核的重要性。将一个普通的CPU专门用于这一目的是对硅和电力的相当大的浪费。至少,使用GPU是一个节能的解决方案。
然而,通过使用特殊用途的硬件,甚至可以达到更高的效率。下面是一个概述:https://blog.inten.to/hardware-for-deep-learning-part-4-asic-96a542fe6a81 在某种程度上,这些特殊用途的处理器是收缩阵列的再世。
降低精度
见3.7.4.2节。
Stuff
通用近似定理
设$\varphi(⋅)$是一个非常数、有界、单调增加的连续函数。让$𝐼𝑚$表示$𝑚$维的单位超立方体$[0,1]^m$。$𝐼𝑚$上的连续函数空间用$C(I_m)$表示。那么,给定任何函数$f\in C(I_m)$和$\varepsilon>0$,存在一个整数$N$,实常数$v_i,b_i\in \mathbb{R}$和实向量$w_i\in \mathbb{R}^m$,其中$i=1,⋯,N$,这样我们可以定义。
作为函数$f$的近似实现,其中$f$与$\varphi$无关;即。
对于所有$x\in Im$。换句话说,$F(x)$形式的函数在$C(I_m)$中是密集的。
NN可以近似于乘法吗?
https://stats.stackexchange.com/questions/217703/can-deep-neural-network-approximate
传统的神经网络由线性图和Lipschitiz激活函数组成。作为Lischitz连续函数的组合,神经网络也是Lipschitz连续的,但乘法不是Lipschitz连续。这意味着,当$x$或$y$中的一个数字过大时,神经网络工作不能近似于乘法。